의약품 제조업체: Alembic Pharmaceuticals Inc. (Updated: 2023-02-17)
처방 정보 요약
ERLOTINIB 정제, 경구용
최초 미국 승인: 2004
적응증 및 용법
Erlotinib 정제는 다음과 같은 적응증을 갖는 kinase 억제제입니다: (1)
- FDA 승인 검사로 확인된 epidermal growth factor receptor (EGFR) exon 19 결실 또는 exon 21 (L858R) 치환 돌연변이가 있는 종양을 가진 전이성 비소세포폐암(NSCLC) 환자의 1차, 유지, 또는 최소 1회 이상의 이전 화학요법 후 진행에 따른 2차 이상의 치료. (1.1)
- gemcitabine과 병용하여 국소 진행성, 절제 불가능 또는 전이성 췌장암 환자의 1차 치료. (1.2)
사용 제한: (1)
- 다른 EGFR 돌연변이를 가진 NSCLC 환자에서의 erlotinib 정제의 안전성과 유효성은 확립되지 않았습니다. (1.1)
- Erlotinib 정제는 platinum 기반 화학요법과 병용하는 것은 권장되지 않습니다. (1.1)
용법 용량
- NSCLC: 공복에 1일 1회 150mg 경구 투여. (2.2)
- 췌장암: 공복에 1일 1회 100mg 경구 투여. (2.3)
제형 및 함량
정제: 25mg, 100mg, 150mg (3) (3)
금기사항
없음. (4) (4)
경고 및 주의사항
- 간질성 폐질환(ILD): 1.1%의 환자에서 발생. 호흡곤란, 기침, 발열 등 급성 신규 또는 진행성 설명되지 않는 폐 증상이 있는 경우 erlotinib 투여를 보류하십시오. ILD로 진단된 경우 erlotinib 투여를 중단하십시오. (5.1)
- 신부전: 특히 탈수 위험이 있는 환자에서 신기능과 전해질을 모니터링하십시오. 중증 신독성의 경우 erlotinib 투여를 보류하십시오. (5.2)
- 간독성: 간부전 및 간신증후군을 포함하여 간장애 유무에 관계없이 발생: 정기적인 간기능 검사를 모니터링하십시오. 중증 또는 악화되는 간기능 검사 결과의 경우 erlotinib 투여를 보류하거나 중단하십시오. (5.3)
- 위장관 천공: Erlotinib 투여를 중단하십시오. (5.4)
- 수포성 및 박리성 피부 장애: Erlotinib 투여를 중단하십시오. (5.5)
- 뇌혈관 사고(CVA): 췌장암 환자에서 CVA의 위험이 증가합니다. (5.6)
- 미세혈관병성 용혈성 빈혈(MAHA): 췌장암 환자에서 MAHA의 위험이 증가합니다. (5.7)
- 안과적 장애: 각막 천공, 궤양 또는 지속적인 중증 각막염의 경우 erlotinib 투여를 중단하십시오. (5.8)
- Warfarin 복용 환자의 출혈: Warfarin 또는 기타 coumarin 유도체 항응고제를 복용하는 환자에서 INR을 정기적으로 모니터링하십시오. (5.9)
- 배아-태아 독성: 태아에게 유해할 수 있습니다. 가임기 여성에게 태아에 대한 잠재적 위험성을 알리고 효과적인 피임을 사용하도록 조언하십시오. (5.10, 8.1, 8.3)
이상반응
약물 상호작용
- CYP3A4 억제제 또는 CYP3A4와 CYP1A2 억제제의 병용은 erlotinib 혈장 농도를 증가시킵니다. 병용 사용을 피하십시오. 불가능한 경우 erlotinib 용량을 감량하십시오. (2.4, 7)
- CYP3A4 유도제는 erlotinib 혈장 농도를 감소시킵니다. 병용 사용을 피하십시오. 불가능한 경우 erlotinib 용량을 증량하십시오. (2.4, 7)
- 흡연 및 CYP1A2 유도제는 erlotinib 혈장 농도를 감소시킵니다. 병용 사용을 피하십시오. 불가능한 경우 erlotinib 용량을 증량하십시오. (2.4, 7)
- 위산도를 증가시키는 약물은 erlotinib 혈장 농도를 감소시킵니다. 양성자 펌프 억제제의 경우 가능하면 병용 사용을 피하십시오. H-2 수용체 길항제의 경우 H-2 수용체 길항제 투여 10시간 후에 erlotinib을 복용하십시오. 제산제와 함께 사용하는 경우 투여 간격을 여러 시간 띄우십시오. (2.4, 7)
특정 집단에서의 사용
수유: 모유 수유하지 마십시오 (8.2) (8)
환자 상담 정보는 17번을 참조하십시오.
개정: 2023년 2월
목차
전체 처방 정보: 목차*
1 적응증 및 용법
1.1 비소세포폐암 (NSCLC)
1.2 췌장암
2 용량 및 투여 방법
2.1 전이성 NSCLC 환자의 선택
2.2 권장 용량 – NSCLC
2.3 권장 용량 – 췌장암
2.4 용량 조절
3 제형 및 함량
4 금기사항
5 경고 및 주의사항
5.1 간질성 폐질환 (ILD)
5.2 신부전
5.3 간손상을 동반하거나 동반하지 않는 간독성
5.4 위장관 천공
5.5 수포성 및 박리성 피부 질환
5.6 뇌혈관 사고
5.7 혈소판감소증을 동반한 미세혈관병성 용혈성 빈혈
5.8 안과적 장애
5.9 Warfarin을 복용하는 환자에서의 출혈
5.10 태아 독성
6 이상반응
6.1 임상시험 경험
6.2 시판 후 경험
7 약물 상호작용
8 특수 집단에서의 사용
8.1 임신
8.2 수유
8.3 가임기 여성 및 남성
8.4 소아 사용
8.5 노인 사용
8.6 간장애
10 과량 투여
11 제품 설명
12 임상 약리
12.1 작용 기전
12.3 약동학
13 비임상 독성학
13.1 발암성, 변이원성, 생식 능력 장애
14 임상 연구
14.1 비소세포폐암 (NSCLC) – EGFR 돌연변이 환자의 1차 치료
14.2 NSCLC – EGFR 돌연변이가 없는 환자의 유지 요법에서 Erlotinib의 효능 부족
14.3 NSCLC – 유지 치료 또는 2/3차 치료
14.4 NSCLC – 항암화학요법과 병용하여 투여한 Erlotinib의 효능 부족
14.5 췌장암 – Gemcitabine과 병용하여 투여한 Erlotinib
16 공급/보관 및 취급 방법
17 환자 상담 정보
- *
- 전체 처방 정보에 포함되지 않은 섹션 또는 하위 섹션은 나열되지 않았습니다.
1 적응증 및 사용법
1.1 비소세포폐암(NSCLC)
Erlotinib 정제는 다음과 같은 경우에 적응증이 있습니다:
- FDA 승인 검사로 확인된 epidermal growth factor receptor (EGFR) exon 19 결실 또는 exon 21 (L858R) 치환 돌연변이가 있는 전이성 비소세포폐암(NSCLC) 환자의 1차 치료, 유지 요법, 또는 이전에 최소 1회 이상의 화학요법 후 진행된 2차 이상의 치료 [임상시험 참조 (14.1, 14.3)].
사용 제한:
- 다른 EGFR 돌연변이가 있는 NSCLC 환자에서 erlotinib 정제의 안전성과 유효성은 확립되지 않았습니다 [임상시험 참조 (14.1, 14.2)].
- Erlotinib 정제는 platinum 기반 화학요법과 병용하는 것이 권장되지 않습니다 [임상시험 참조 (14.4)].
1.2 췌장암
Gemcitabine과 병용투여 시 erlotinib 정제는 국소 진행성, 절제 불가능 또는 전이성 췌장암 환자의 1차 치료에 적응증이 있습니다 [임상시험 참조 (14.5)].
2 용량 및 투여
2.1 전이성 NSCLC 환자의 선택
종양 또는 혈장 검체에서 EGFR exon 19 결실 또는 exon 21 (L858R) 치환 돌연변이의 존재 여부에 따라 erlotinib 정제로 전이성 NSCLC 치료 환자를 선택합니다 [임상 연구 (14.1, 14.2) 참조]. 혈장 검체에서 이러한 돌연변이가 검출되지 않는 경우 가능하다면 종양 조직을 검사하십시오. NSCLC의 EGFR 돌연변이 검출을 위한 FDA 승인 검사에 대한 정보는 http://www.fda.gov/CompanionDiagnostics에서 확인할 수 있습니다.
2.2 권장 용량 – NSCLC
NSCLC에 대한 erlotinib 정제의 권장 일일 용량은 공복에 150 mg을 복용하는 것입니다. 즉, 음식 섭취 최소 1시간 전 또는 2시간 후에 복용합니다. 질병이 진행되거나 허용할 수 없는 독성이 발생할 때까지 치료를 계속해야 합니다.
2.3 권장 용량 – 췌장암
췌장암에 대한 erlotinib 정제의 권장 일일 용량은 gemcitabine과 병용하여 하루에 한 번 100mg을 복용하는 것입니다. Erlotinib 정제는 공복에 복용해야 합니다. 즉, 음식 섭취 최소 1시간 전 또는 2시간 후에 복용합니다. 질병이 진행되거나 허용할 수 없는 독성이 발생할 때까지 치료를 계속해야 합니다 [임상 연구 (14.5) 참조].
2.4 용량 조정
| 부작용 |
||
| 폐† |
간질성 폐렴(ILD) | 엘로티닙 정제 중단 |
| 가능한 ILD에 대한 진단 평가 중 | 엘로티닙 정제 보류* |
|
| 간† |
3주 이내에 현저히 개선되거나 해결되지 않는 중증 간독성 | 엘로티닙 정제 중단 |
| 기존 간장애 또는 담즙 폐쇄가 있는 환자에서 기저치의 2배 빌리루빈 또는 3배 트란스아미나제 수치 증가 | 엘로티닙 정제 보류* 및 중단 고려 | |
| 기존 간장애가 없는 환자에서 총 빌리루빈 수치가 정상 상한치의 3배 이상 또는 트란스아미나제가 정상 상한치의 5배 이상 | 엘로티닙 정제 보류* 및 중단 고려 | |
| 신장† |
중증(CTCAE 3~4등급) 신독성 | 엘로티닙 정제 보류* 및 중단 고려 |
| 위장관† |
위장관 천공 | 엘로티닙 정제 중단 |
| 약물 관리에 반응하지 않는 지속적인 중증 설사(예: 로페라미드) | 엘로티닙 정제 보류* |
|
| 피부† |
중증 수포, 물집 또는 박리성 피부 질환 | 엘로티닙 정제 중단 |
| 약물 관리에 반응하지 않는 중증 발진 | 엘로티닙 정제 보류* |
|
| 안과† |
각막 천공 또는 중증 궤양 | 엘로티닙 정제 중단 |
| 각막염(NCI-CTC 버전 4.0) 3~4등급 또는 2등급이 2주 이상 지속 | 엘로티닙 정제 보류* |
|
| 급성/악화된 안과적 장애(예: 안구통) | 엘로티닙 정제 보류* 및 중단 고려 | |
| 약물 상호작용 |
||
| CYP3A4 억제제‡ |
강력한 CYP3A4 억제제[아타자나비르, 클래리스로마이신, 인디나비르, 이트라코나졸, 케토코나졸, 네파조돈, 넬피나비르, 리토나비르, 사퀴나비르, 텔리스로마이신, 트롤레안도마이신(TAO), 보리코나졸 또는 그레이프푸르츠나 그레이프푸르츠 주스] 또는 CYP3A4 및 CYP1A2 모두의 억제제(예: 시프로플록사신)와 병용 시 중증 반응이 발생하면 | 엘로티닙 정제 50mg씩 감량, 가능하면 병용 피함 |
| CYP3A4 유도제‡ |
CYP3A4 유도제(리팜피신, 리파부틴, 리파펜틴, 페니토인, 카르바마제핀, 페노바르비탈 또는 St. John’s Wort) 병용 | 최대 450mg까지 내약성 한도 내 2주마다 50mg씩 증량, 가능하면 병용 피함 |
| 동시 흡연 ‡§ |
동시 흡연 | 최대 300mg까지 2주마다 50mg씩 증량, 금연 시 즉시 엘로티닙 정제 용량을 권장량(150mg 또는 100mg 1일 1회)으로 감량 |
| 프로톤 펌프 억제제 | 분할 투여 시 프로톤 펌프 억제제가 상부 위장관 pH에 장시간 영향을 주기 때문에 상호작용 제거 불가능 | 가능하면 병용 피함 |
| H2-수용체 길항제 | 라니티딘과 같은 H2-수용체 길항제 병용이 필요한 경우 투여 시간 간격을 둠 | 엘로티닙 정제는 H2-수용체 길항제 투여 10시간 후 및 다음 H2-수용체 길항제 투여 2시간 전에 복용 |
| 제산제 | 제산제가 엘로티닙 약동학에 미치는 영향은 평가되지 않음 | 제산제가 필요한 경우 제산제 및 엘로티닙 정제 투여 시간을 수 시간 간격으로 분리 |
† 자세한 내용은 경고 및 주의사항(5)을 참조.
* 용량 제한 독성이 기준치 또는 grade ≤ 1로 해결된 후 치료를 재개할 때는 erlotinib tablets을 50 mg 단위로 감량합니다.
‡ 추가 정보는 Drug Interactions (7)을 참조하십시오.
§ 추가 정보는 Clinical Pharmacology (12.3)을 참조하십시오.
3 제형 및 함량
Erlotinib 정제는 다음과 같은 함량으로 제공됩니다:
Erlotinib 정제 25 mg은 원형의 양면이 볼록한 흰색 필름코팅정제로서 한 면에는 “L55″가 음각되어 있고 다른 면은 평평합니다.
Erlotinib 정제 100 mg은 원형의 양면이 볼록한 흰색 필름코팅정제로서 한 면에는 “L630″이 음각되어 있고 다른 면은 평평합니다.
Erlotinib 정제 150 mg은 원형의 양면이 볼록한 흰색 필름코팅정제로서 한 면에는 “L631″이 음각되어 있고 다른 면은 평평합니다.
4 금기사항
없음.
5 경고 및 주의사항
5.1 간질성 폐질환 (ILD)
에를로티닙 치료 중 심각한 ILD 사례, 치명적인 사례를 포함하여 발생할 수 있습니다. 비조절 연구 및 동시 화학요법을 시행한 약 32,000명의 에를로티닙 치료 환자에서 ILD의 전반적인 발생률은 약 1.1%였습니다. ILD 환자에서 증상 발현은 에를로티닙 치료 시작 후 5일에서 9개월 이상(중앙값 39일) 사이에 나타났습니다.
호흡곤란, 기침, 발열 등 호흡기 증상이 급성으로 발생할 경우 에를로티닙 투여를 중단하고 진단 평가를 위해 기다리십시오. ILD가 확인되면 에를로티닙을 영구 중단하십시오 [용량 및 투여법(2.4) 참조].
5.2 신부전
에를로티닙 치료 중 간신증후군, 심한 급성 신부전(치명적인 사례 포함) 및 신기능부전이 발생할 수 있습니다. 신부전은 기저 간 기능 장애 또는 심한 탈수로 인해 발생할 수 있습니다. 3개의 단일요법 폐암 연구에서 심한 신기능부전의 발생률은 에를로티닙군에서 0.5%, 대조군에서 0.8%였습니다. 췌장암 연구에서 신기능부전의 발생률은 에를로티닙 플러스 젬시타빈군에서 1.4%, 대조군에서 0.4%였습니다. 심한 신기능부전이 발생한 환자에게는 신독성이 해소될 때까지 에를로티닙을 중단하십시오. 에를로티닙 치료 중 신기능 및 혈청 전해질 주기적인 모니터링을 시행하십시오 [부작용(6.1) 및 용량 및 투여법(2.4) 참조].
5.3 간독성 및 간기능 장애 여부에 관계없는 간독성
정상 간 기능을 가진 환자에서도 에를로티닙 치료 중 간기능 장애 및 간신증후군(치명적인 사례 포함)이 발생할 수 있으며, 기저 간 기능 장애 환자에서 간독성 위험이 증가합니다. 중증 간 기능 장애 환자가 배제된 임상 연구에서 3개의 단일요법 폐암 연구에서 간실패의 발생률은 에를로티닙군에서 0.4%, 대조군에서 0%였습니다. 췌장암 연구에서 간실패의 발생률은 에를로티닙 플러스 젬시타빈군에서 0.4%, 대조군에서 0.4%였습니다. 중증 간 기능 장애(Child-Pugh B)를 가진 15명의 환자를 대상으로 한 약동학 연구에서, 이 중 15명의 환자 중 10명이 마지막 에를로티닙 투여 후 30일 이내에 사망했습니다. 이 중 1명은 간신증후군으로 사망하였으며, 1명은 급속한 간기능 장애로 사망하였으며, 나머지 8명은 진행성 질환으로 사망하였습니다. 사망한 10명 중 6명은 기저 총빌리루빈 수치가 정상 상한치의 3배 이상이었습니다.
에를로티닙 치료 중 간기능 검사(전이아미노전이효소, 빌리루빈 및 알칼리성 인산효소)를 주기적으로 시행하십시오. 기저 간 기능 장애나 담관 폐색을 가진 환자에게는 총빌리루빈 수치가 정상 상한치의 3배 이상이거나 전이아미노전이효소 수치가 정상 상한치의 5배 이상인 경우 간기능 주기적인 모니터링을 더 자주 시행하십시오. 기저 간 기능 장애나 담관 폐색을 가진 환자에게는 기저치에 비해 빌리루빈이 2배 증가하거나 전이아미노전이효소 수치가 기저치 대비 3배 증가한 경우 에를로티닙을 중단하십시오. 위의 기준을 충족하지 않는 이상적인 간기능 검사 결과가 3주 이내에 개선되지 않거나 해소되지 않는 환자에게는 에를로티닙을 중단하십시오 [용량 및 투여법(2.4) 및 임상약리학(12.3) 참조].
5.4 위장관 천공
에를로티닙 치료 중 위장관 천공(치명적인 사례 포함)이 발생할 수 있습니다. 혈관신생억제제, 스테로이드, 비스테로이드성 항염증제, 또는 탁사인 기반 화학요법을 동시에 투여받는 환자 또는 궤양성궤양 또는 충수질환의 이력이 있는 환자는 천공 발생 위험이 증가할 수 있습니다 [부작용(6.1, 6.2) 참조]. 3개의 단일요법 폐암 연구에서 위장관 천공의 발생률은 에를로티닙군에서 0.2%, 대조군에서 0.1%였습니다. 췌장암 연구에서 위장관 천공의 발생률은 에를로티닙 플러스 젬시타빈군에서 0.4%, 대조군에서 0%였습니다. 위장관 천공이 발생한 환자에게는 에를로티닙을 영구 중단하십시오 [용량 및 투여법(2.4) 참조].
5.5 불루스 및 탈피성 피부 장애
에를로티닙 치료 중 불루스, 수포성 피부 장애 및 탈피성 피부 장애(Stevens-Johnson 증후군/중독성 표피괴사를 시사하는 사례 포함)가 발생할 수 있습니다 [부작용(6.1, 6.2) 참조]. 3개의 단일요법 폐암 연구에서 불루스 및 탈피성 피부 장애의 발생률은 에를로티닙군에서 1.2%, 대조군에서 0%였습니다. 췌장암 연구에서 불루스 및 탈피성 피부 장애의 발생률은 에를로티닙 플러스 젬시타빈군에서 0.4%, 대조군에서 0%였습니다. 심한 불루스, 수포성 또는 탈피성 피부 장애가 발생한 환자에게는 에를로티닙 치료를 중단하십시오 [용량 및 투여법(2.4) 참조].
5.6 뇌혈관 사고
췌장암 임상시험에서 에를로티닙/젬시타빈 군에서 뇌혈관 사고가 7명(발생률: 2.5%) 발생했습니다. 이 중 1명은 출혈성이며 유일한 치명적인 사건이었습니다. 대조군에서는 뇌혈관 사고가 없었습니다. 3개의 단일요법 폐암 연구에서 뇌혈관 사고의 발생률은 에를로티닙군에서 0.6%로 대조군에서 관찰된 수준보다 높지 않았습니다.
5.7 혈소판감소증을 동반한 미세혈관성 용혈성 빈혈
3개의 단일요법 폐암 연구에서 미세혈관성 용혈성 빈혈을 동반한 혈소판감소증의 발생률은 에를로티닙군에서 0%, 대조군에서 0.1%였습니다. 췌장암 연구에서 미세혈관성 용혈성 빈혈을 동반한 혈소판감소증의 발생률은 에를로티닙 플러스 젬시타빈군에서 1.4%, 대조군에서 0%였습니다.
5.8 안구 장애
에를로티닙 치료 중 눈물 생성 감소, 이상한 속눈썹 성장, 건조각막염 또는 각막염이 발생할 수 있으며, 각막 천공 또는 궤양으로 이어질 수 있습니다 [부작용(6.1) 및 (6.2) 참조]. 3개의 단일요법 폐암 연구에서 안구 장애의 발생률은 에를로티닙군에서 17.8%, 대조군에서 4%였습니다. 췌장암 연구에서 안구 장애의 발생률은 에를로티닙 플러스 젬시타빈군에서 12.8%, 대조군에서 11.4%였습니다. 급성 또는 악화되는 안구 장애(눈 통증 등)가 발현된 환자에게는 에를로티닙 치료를 중단하거나 중단하십시오 [용량 및 투여법(2.4) 참조].
5.9 와파린 복용 환자에서의 출혈
에를로티닙과 와파린을 동시에 투여하는 경우 국제표준화율(INR) 상승과 관련된 심한 출혈 및 치명적인 출혈이 발생할 수 있습니다. 와파린 또는 다른 쿠마린 유도체 항응고제를 복용하는 환자에게는 에를로티닙 치료 중 프로트롬빈 시간과 INR을 정기적으로 모니터링하십시오 [부작용(6.1) 및 약물 상호작용(7) 참조].
5.10 배아-태아 독성
동물 자료 및 작용 기전에 따르면, 에를로티닙은 임신한 여성에게 투여될 경우 태아에 유해를 일으킬 수 있습니다. 장기형성기에 투여할 경우, 에를로티닙 투여는 토끼에서 권장 인체 일일 투여량의 약 3배 노출에서 배아-태아 사망 및 유산을 유발했습니다. 임신한 여성에게 태아에 대한 잠재적 위험을 알려주십시오.
치료 중 및 마지막 투여 후 1개월 동안 생식 가능한 여성에게는 효과적인 피임을 사용할 것을 권장하십시오 [특정 인구에 대한 사용(8.1) 및 (8.3), 임상약리학(12.1) 참조].
6 부작용 반응
아래와 같은 심각한 부작용은 사망을 포함할 수 있으며, labeling의 다른 섹션에서 더 자세히 다루어집니다:
- Interstitial Lung Disease (ILD) [Warnings and Precautions (5.1) 참조]
- 신부전 [Warnings and Precautions (5.2) 참조]
- 간 장애 동반 또는 비동반 간독성 [Warnings and Precautions (5.3) 참조]
- 위장관 천공 [Warnings and Precautions (5.4) 참조]
- Bullous and Exfoliative Skin Disorders [Warnings and Precautions (5.5) 참조]
- 뇌졸중 [Warnings and Precautions (5.6) 참조]
- Microangiopathic Hemolytic Anemia with Thrombocytopenia [Warnings and Precautions (5.7) 참조]
- 안과적 장애 [Warnings and Precautions (5.8) 참조]
- Warfarin 복용 환자에서의 출혈 [Warnings and Precautions (5.9) 참조]
6.1 임상시험 경험
임상시험은 매우 다양한 조건에서 수행되므로 한 약물의 임상시험에서 관찰된 이상반응 발생률을 다른 약물의 임상시험에서의 발생률과 직접 비교할 수 없으며 실제로 관찰되는 발생률을 반영하지 않을 수 있습니다.
Erlotinib 안전성 평가는 1200명 이상의 암 환자를 대상으로 erlotinib 단독요법, 300명 이상의 환자를 대상으로 erlotinib 100mg 또는 150mg과 gemcitabine 병용요법, 1228명의 환자를 대상으로 erlotinib과 다른 항암제 병용요법으로 수행되었습니다. Erlotinib에서 가장 흔한 이상반응은 발진과 설사로 보통 치료 첫 달 이내에 발생합니다. NSCLC와 췌장암 치료를 위한 erlotinib 임상연구에서 발진과 설사의 발생률은 각각 70%, 42%였습니다.
비소세포폐암
EGFR 돌연변이 환자의 1차 치료
Erlotinib 치료 환자에서 가장 빈번한(≥ 30%) 이상반응은 설사, 무력증, 발진, 기침, 호흡곤란, 식욕감퇴였습니다. Erlotinib 치료 환자에서 발진 발생까지의 중앙값은 15일, 설사 발생까지의 중앙값은 32일이었습니다.
Erlotinib 치료 환자에서 가장 빈번한 3-4등급 이상반응은 발진과 설사였습니다.
Erlotinib 치료 환자의 37%에서 이상반응으로 인한 용량 중단 또는 감량이 발생했고, 14.3%의 환자는 이상반응으로 인해 치료를 중단했습니다. Erlotinib 치료 환자에서 용량 조절을 유발한 가장 빈번한 이상반응은 발진(13%), 설사(10%), 무력증(3.6%)이었습니다.
연구 1에서 erlotinib 또는 화학요법을 받은 환자의 최소 10%에서 발생하고 erlotinib 치료군에서 ≥ 5% 증가한 흔한 이상반응은 National Cancer Institute Common Toxicity Criteria for Adverse Events version 3.0 (NCI-CTCAE v3.0) 등급으로 표 1에 기재되어 있습니다. 연구 1에서 erlotinib 치료 기간의 중앙값은 9.6개월이었습니다.
표 1: 발생률 ≥ 10% 이상이고 Erlotinib 치료군에서 ≥ 5% 증가한 이상반응(연구 1)
| 부작용 |
Erlotinib N = 84 |
화학요법† N = 83 |
||
| 모든 등급 % |
3-4 등급 % |
모든 등급 % |
3-4 등급 % |
|
| 발진‡ |
85 | 14 | 5 | 0 |
| 설사 | 62 | 5 | 21 | 1 |
| 기침 | 48 | 1 | 40 | 0 |
| 호흡곤란 | 45 | 8 | 30 | 4 |
| 건조한 피부 | 21 | 1 | 2 | 0 |
| 허리통증 | 19 | 2 | 5 | 0 |
| 가슴통증 | 18 | 1 | 12 | 0 |
| 결막염 | 18 | 0 | 0 | 0 |
| 점막 염증 | 18 | 1 | 6 | 0 |
| 가려움증 | 16 | 0 | 1 | 0 |
| 구순구각 염 | 14 | 0 | 0 | 0 |
| 관절통 | 13 | 1 | 6 | 1 |
| 근골격계 통증 |
11 | 1 | 1 | 0 |
† 백금 기반 화학요법 (시스플라틴 또는 카보플라틴과 젬시타빈 또는 docetaxel).
‡ 발진은 발진, 여드름, 모낭염, 홍반, 여드름성 피부염, 피부염, 수족증후군, 박리성 발진, 홍반성 발진, 가려운 발진, 피부 독성, 습진, 모낭 발진, 피부 궤양을 포함한 복합 용어입니다.
간 독성: 연구 1에서 erlotinib 치료 환자 1명이 치명적인 간 부전을 경험했고 4명의 추가 환자는 3-4등급의 간기능 검사 이상을 경험했습니다 [경고 및 주의사항 (5.3) 참조].
유지 요법
무작위 유지 요법 시험(연구 3)에서 단일 요법 erlotinib 150mg를 투여받은 환자들에게 발생한 원인과 관계없이 3% 이상 발생하고 위약군보다 3% 이상 더 많이 발생한 부작용은 NCI-CTCAE v3.0 등급별로 표 2에 요약되어 있습니다.
단일 요법 erlotinib 150mg를 투여받은 환자에게 가장 흔한 부작용은 발진과 설사였습니다. 3-4등급 발진과 설사는 각각 erlotinib 치료 환자의 9%와 2%에서 발생했습니다. 발진과 설사로 인해 각각 1%와 0.5%의 환자가 연구를 중단했습니다. 발진과 설사로 인해 각각 5%와 3%의 환자에서 용량 감량 또는 투약 중단이 필요했습니다. Erlotinib 치료 환자에서 발진 발생까지의 중앙값은 10일, 설사 발생까지의 중앙값은 15일이었습니다.
표 2: NSCLC 유지 요법 연구: 단일 요법 Erlotinib 군에서 위약군에 비해 10% 이상의 발생률 증가와 5% 이상의 발생률 증가를 보인 부작용 (연구 3)
| 부작용 |
Erlotinib N = 433 |
위약 N = 445 |
||||
| 모든 등급 |
3등급 |
4등급 |
모든 등급 |
3등급 |
4등급 |
|
| % |
% |
% |
% |
% |
% |
|
| 발진† |
60 | 9 | 0 | 9 | 0 | 0 |
| 설사 | 20 | 2 | 0 | 4 | 0 | 0 |
†발진은 복합어로서 발진, 여드름, 여드름성 피부염, 피부 갈라짐, 홍반, 구진성 발진, 전신 발진, 가려운 발진, 피부 박리, 두드러기, 피부염, 습진, 박리성 발진, 박리성 피부염, 종기, 반점 발진, 농포성 발진, 피부 과색소침착, 피부 반응, 피부 궤양을 포함합니다.
ALT 상승을 포함한 간기능 검사 이상이 erlotinib 투여 환자의 3%와 위약 투여 환자의 1%에서 2등급 이상의 중증도로 관찰되었습니다. 2등급 이상의 빌리루빈 상승은 erlotinib 투여 환자의 5%와 위약군의 1% 미만에서 관찰되었습니다 [용법 및 투여량(2.4) 및 경고 및 주의사항(5.3) 참조].
2차/3차 치료
비소세포폐암 환자를 대상으로 한 무작위 시험에서 단일제 erlotinib 150mg 투여군과 위약군 간에 10% 이상의 발생 빈도 차이가 있었고 발생 빈도가 최소 5% 높은 이상반응은 NCI-CTC v2.0 등급별로 요약되어 표 3에 나타납니다.
이 환자군에서 가장 흔한 이상반응은 발진과 설사였습니다. 3-4등급 발진과 설사의 발생률은 각각 erlotinib 투여 환자의 9%, 6%였습니다. 발진과 설사로 인한 투여 중단률은 각각 1%였습니다. 발진과 설사로 인한 용량 감량이 필요했던 환자는 각각 6%와 1%였습니다. 발진과 설사의 중앙 발생 시기는 각각 8일과 12일이었습니다.
표 3: NSCLC 2차/3차 치료 연구: 단일제 Erlotinib 투여군에서 위약군 대비 10% 이상 발생하고 5% 이상 높은 이상반응 (연구 4)
| 부작용 |
엘로티닙 150 mg N=485 |
위약 N=242 |
||||
| 모든 등급 |
3등급 |
4등급 |
모든 등급 |
3등급 |
4등급 |
|
| % |
% |
% |
% |
% |
% |
|
| 발진† |
75 | 8 | <1 | 17 | 0 | 0 |
| 설사 | 54 | 6 | <1 | 18 | <1 | 0 |
| 식욕부진 | 52 | 8 | 1 | 38 | 5 | <1 |
| 피로 | 52 | 14 | 4 | 45 | 16 | 4 |
| 호흡곤란 | 41 | 17 | 11 | 35 | 15 | 11 |
| 구역질 | 33 | 3 | 0 | 24 | 2 | 0 |
| 감염 | 24 | 4 | 0 | 15 | 2 | 0 |
| 구내염 | 17 | <1 | 0 | 3 | 0 | 0 |
| 가려움증 | 13 | <1 | 0 | 5 | 0 | 0 |
| 건조 피부 | 12 | 0 | 0 | 4 | 0 | 0 |
| 결막염 | 12 | <1 | 0 | 2 | <1 | 0 |
| 건조안구증후군 | 12 | 0 | 0 | 3 | 0 | 0 |
†발진은 발진, 반고리 수장구진성 홍반, 여드름, 피부 장애, 색소 침착 장애, 홍반, 피부궤양, 박탈피부염, 구진성 발진, 피부박리를 포함하는 복합 용어입니다.
단독요법으로 erlotinib 150 mg을 투여받은 환자에서 간 기능 검사 이상(alanine aminotransferase (ALT), aspartate aminotransferase (AST) 및 bilirubin 상승 포함)이 관찰되었습니다. 이러한 상승은 주로 일시적이거나 간 전이와 관련이 있었습니다. 2등급 [정상 상한치(ULN)의 2.5 ~ 5배]의 ALT 상승은 erlotinib 투여 환자의 4%, 위약 투여 환자의 1% 미만에서 발생했습니다. Erlotinib 투여 환자에서는 3등급(ULN의 5 ~ 20배) 상승이 관찰되지 않았습니다. 간 기능 변화가 심각한 경우 erlotinib 투여를 중단하거나 중지해야 합니다 [용법 용량(2.4) 참조].
췌장암-Gemcitabine과 병용 투여한 Erlotinib
이 연구는 국소 진행성, 절제 불가능 또는 전이성 췌장암 환자를 대상으로 erlotinib(150 mg 또는 100 mg 1일 1회) 또는 위약과 gemcitabine(1000 mg/m2, 정맥 주입)을 병용 투여한 무작위, 이중 눈가림, 위약 대조 연구였습니다(연구 5). 안전성 인구는 erlotinib군(100 mg 코호트 259명, 150 mg 코호트 23명)에서 282명, 위약군(100 mg 코호트 256명, 150 mg 코호트 24명)에서 280명이었습니다.
췌장암 환자 대상 무작위 시험(연구 5)에서 gemcitabine과 함께 erlotinib 100 mg을 투여받은 환자의 10% 이상에서 발생한 이상반응은 NCI-CTC v2.0에 따라 표 4와 같이 등급화되었습니다.
Gemcitabine과 함께 erlotinib 100 mg을 투여받은 췌장암 환자에서 가장 흔한 이상반응은 피로, 발진, 오심, 식욕부진, 설사였습니다. Erlotinib과 gemcitabine 병용 투여군에서 3 ~ 4등급 발진과 설사가 각각 5%에서 보고되었습니다. 발진과 설사 발생까지의 중간 시간은 각각 10일과 15일이었습니다. 발진과 설사는 각각 2%의 환자에서 용량 감소를 초래했고, erlotinib과 gemcitabine을 병용 투여받은 환자의 최대 1%에서 연구 중단을 초래했습니다. Erlotinib과 gemcitabine 병용 투여군에서 5% 미만의 발생률을 보인 중대한 이상반응(NCI-CTC 3등급 이상)으로는 실신, 부정맥, 장폐색증, 췌장염, 혈소판감소증을 동반한 미세혈관병성 용혈성 빈혈을 포함한 용혈성 빈혈, 심근경색/허혈, 뇌출혈을 포함한 뇌혈관 사고, 신부전 등이 있었습니다 [경고 및 주의사항(5) 참조].
150 mg 코호트는 발진과 같은 특정 약물 종류 관련 이상반응의 더 높은 비율과 연관이 있었고 더 자주 용량 감량이나 투여 중단이 필요했습니다.
표 4: 발생률 10% 이상이고 Erlotinib 투여 췌장암 환자에서 5% 이상 증가한 이상반응: 100 mg 코호트(연구 5)
| 부작용 |
엘로티닙 + 젬시타빈 1000 mg/m2 정맥주사 N=259 |
위약 + 젬시타빈 1000 mg/m2 정맥주사 N=256 |
||||
|---|---|---|---|---|---|---|
| 모든 등급 |
3등급 |
4등급 |
모든 등급 |
3등급 |
4등급 |
|
| % |
% |
% |
% |
% |
% |
|
| 발진† |
70 | 5 | 0 | 30 | 1 | 0 |
| 설사 | 48 | 5 | <1 | 36 | 2 | 0 |
| 체중 감소 | 39 | 2 | 0 | 29 | <1 | 0 |
| 감염* |
39 | 13 | 3 | 30 | 9 | 2 |
| 발열 | 36 | 3 | 0 | 30 | 4 | 0 |
| 구내염 | 22 | <1 | 0 | 12 | 0 | 0 |
| 우울증 | 19 | 2 | 0 | 14 | <1 | 0 |
| 기침 | 16 | 0 | 0 | 11 | 0 | 0 |
| 두통 | 15 | <1 | 0 | 10 | 0 | 0 |
* 감염은 불특정 병원체 감염뿐만 아니라 세균(클라미디아, 리켓치아, 마이코박테리아 및 마이코플라스마 포함), 기생충(선충, 외부 기생충 및 원충 포함), 바이러스 및 진균 감염을 포함하는 복합 용어입니다.
† 발진은 발진, 손발구 증후군, 색소 침착 장애, 여드름성 피부염, 모낭염, 광과민 반응, 스티븐스-존슨 증후군, 두드러기, 홍반성 발진, 피부 장애, 피부 궤양을 포함하는 복합 용어입니다.
엘로티닙/젬시타빈 그룹의 10명(4%)과 위약/젬시타빈 그룹의 3명(1%)의 환자에게 심부정맥혈전증이 발생했습니다. 심부정맥혈전증을 포함한 3등급 이상의 혈전증 전체 발생률은 엘로티닙 플러스 젬시타빈 그룹에서 11%, 위약 플러스 젬시타빈 그룹에서 9%였습니다.
연구 5에서 간기능 이상(≥ 2등급) 발생률은 표 5에 제시되어 있습니다[용법·용량(2.4) 및 주의사항(5.3)을 참조하세요].
표 5: 췌장암 환자의 간기능 이상: 100 mg 코호트(연구 5)
| Erlotinib + Gemcitabine 1000 mg/m2 IV N=259 |
위약 + Gemcitabine 1000 mg/m2 IV N=256 |
|||||
| 등급 2 |
등급 3 |
등급 4 |
등급 2 |
등급 3 |
등급 4 |
|
| Bilirubin | 17% | 10% | <1% | 11% | 10% | 3% |
| ALT | 31% | 13% | <1% | 22% | 9% | 0% |
| AST | 24% | 10% | <1% | 19% | 9% | 0% |
비소세포폐암 및 췌장 적응증: 선택된 낮은 빈도의 부작용
위장관 장애
일부 와파린 또는 NSAID 병용 투여와 관련된 위장관 출혈(치명적인 경우 포함) 사례가 보고되었습니다 [경고 및 주의사항(5.9) 및 약물상호작용(7) 참조]. 이러한 부작용은 소화성 궤양 출혈(위염, 위십이지장 궤양), 혈제, 혈변, 흑색변 및 가능한 대장염에서 발생한 출혈로 보고되었습니다.
6.2 시판 후 경험
다음 부작용은 Erlotinib의 승인 후 사용 중에 확인되었습니다. 이러한 반응은 크기가 불확실한 인구 집단에서 자발적으로 보고되므로 빈도를 신뢰성 있게 추정하거나 약물 노출과의 인과 관계를 확립하는 것이 항상 가능한 것은 아닙니다.
근골격계 및 결합 조직 장애: 스타틴 요법과 병용 시 횡문근융해증을 포함한 근육병증
안과 장애: 포도막염을 포함한 눈 염증
7 약물 상호작용
CYP3A4 억제제
Erlotinib과 강력한 CYP3A4 억제제 또는 CYP3A4와 CYP1A2 억제제를 병용투여하면 erlotinib 노출이 증가합니다. Erlotinib은 주로 CYP3A4에 의해 대사되고 적은 정도로 CYP1A2에 의해 대사됩니다. Erlotinib 노출 증가는 노출 관련 독성의 위험을 증가시킬 수 있습니다[임상약리학(12.3) 참조].
Erlotinib과 강력한 CYP3A4 억제제(예: boceprevir, clarithromycin, conivaptan, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, telithromycin, voriconazole, 자몽 또는 자몽 주스) 또는 CYP3A4와 CYP1A2 억제제(예: ciprofloxacin)와의 병용투여를 피해야 합니다. 병용투여가 불가피한 경우 강력한 CYP3A4 억제제 또는 CYP3A4와 CYP1A2 억제제와 병용투여 시 erlotinib 용량을 감량하십시오[용법용량(2.4) 참조].
CYP3A4 유도제
Erlotinib 투여 전 CYP3A4 유도제로 전처치하면 erlotinib 노출이 감소합니다[임상약리학(12.3) 참조]. CYP3A4 유도제(예: carbamazepine, phenytoin, rifampin, rifabutin, rifapentine, phenobarbital, St. John’s wort)와의 병용투여가 불가피한 경우 erlotinib 용량을 증량하십시오[용법용량(2.4) 참조].
CYP1A2 유도제와 흡연
흡연은 erlotinib 노출을 감소시킵니다. 담배 흡연(CYP1A2 유도제)을 피하고 중등도 CYP1A2 유도제(예: teriflunomide, rifampin, phenytoin)와 erlotinib의 병용 사용을 피하십시오. 담배를 피우는 환자 또는 중등도 CYP1A2 유도제와의 병용투여가 불가피한 경우 erlotinib 용량을 증량하십시오[용법용량(2.4), 임상약리학(12.3) 참조].
위 pH를 증가시키는 약물
Erlotinib과 프로톤 펌프 억제제(예: omeprazole) 및 H-2 수용체 길항제(예: ranitidine)의 병용투여는 erlotinib 노출을 감소시킵니다[임상약리학(12.3) 참조]. 프로톤 펌프 억제제의 경우 가능하면 병용 사용을 피하십시오. H-2 수용체 길항제 및 제산제의 경우 용법을 조정하십시오[용법용량(2.4) 참조]. 위 pH를 높이는 약제와 병용 투여 시 erlotinib 용량을 증량하는 것은 노출 손실을 보상하기에 충분하지 않을 것입니다.
항응고제
Warfarin을 포함한 쿠마린 유도체 항응고제와의 상호작용으로 INR(International Normalized Ratio) 증가 및 출혈 이상반응이 보고되었으며, 일부 사례에서는 치명적이었습니다. Erlotinib을 투여받는 환자에서 정기적으로 프로트롬빈 시간 또는 INR을 모니터링하십시오. Erlotinib의 용량 조절은 권장되지 않습니다[경고 및 주의사항(5.9), 이상반응(6.1) 참조].
8 특정 집단에서의 사용
8.1 임신
위험 요약
동물 데이터와 작용 기전에 근거할 때, erlotinib은 임신한 여성에게 투여 시 태아에게 유해할 수 있습니다. 임신 중 erlotinib 사용에 대한 제한적인 데이터로는 주요 선천성 기형 또는 유산의 위험을 알리기에 불충분합니다. 기관 형성 기간 동안 erlotinib을 투여하면 권장 인체 1일 투여량 150mg의 약 3배 노출 시 토끼에서 배아-태아 치사 및 유산이 나타났습니다. 임신부에게 태아에 대한 잠재적 위험을 알려야 합니다.
미국 일반 인구에서 임상적으로 인지된 임신에 대한 주요 선천성 기형 및 유산의 추정 배경 위험은 각각 2~4% 및 15~20%입니다.
데이터
동물 데이터
Erlotinib은 기관 형성 기간 동안 인체 권장량에서 달성되는 혈장 약물 농도의 약 3배에 해당하는 용량으로 투여 시 토끼에서 모체 독성으로 인한 배아-태아 치사 및 유산을 일으키는 것으로 나타났습니다(150mg 1일 용량에서의 AUC). 동일 기간 동안 인체 권장 1일 용량에서의 노출과 대략 동등한 용량으로 토끼 또는 랫드에서 배아-태아 치사 또는 유산의 발생률 증가는 없었습니다. 독립적인 수태능 연구에서 30 mg/m2/day 또는 60 mg/m2/day(mg/m2 기준으로 권장 1일 용량의 0.3 또는 0.7배)의 erlotinib으로 치료받은 암컷 랫드는 초기 재흡수가 증가하여 생존 태아 수가 감소했습니다.
토끼에서 150mg/day에서 인체에서 관찰되는 혈장 약물 농도의 3배까지, 랫드에서 60 mg/m2/day(mg/m2 기준 150mg/day의 권장량의 0.7배)까지 기관 형성 기간 동안 erlotinib을 투여한 토끼 또는 랫드에서 최기형성 효과는 관찰되지 않았습니다.
8.2 수유
위험 요약
Erlotinib이 모유에 존재하는지 또는 erlotinib이 모유 수유 유아에 미치는 영향 또는 모유 생성에 미치는 영향에 대한 데이터는 없습니다. 간질성 폐질환, 간독성, 수포성 및 박리성 피부 장애, 미세혈관병성 용혈성 빈혈과 혈소판감소증, 안과 장애, 설사 등 erlotinib으로 인한 심각한 이상반응의 가능성 때문에, 수유부에게 erlotinib 치료 중 및 최종 투여 후 2주 동안 모유 수유를 하지 않도록 권고합니다.
8.3 가임기 여성 및 남성
피임
여성
Erlotinib은 임신한 여성에게 투여 시 태아에게 유해할 수 있습니다[특정 집단에서의 사용(8.1) 참조]. 가임기 여성에게 erlotinib 치료 중 및 erlotinib 최종 투여 후 1개월 동안 효과적인 피임법을 사용하도록 권고합니다.
8.4 소아 사용
소아 환자에서의 erlotinib의 안전성과 유효성은 확립되지 않았습니다.
재발성 또는 불응성 상의세포종 소아 환자 25명(중앙값 14세, 범위 3~20세)을 대상으로 한 공개, 다기관 시험에서 erlotinib 또는 etoposide에 1:1로 무작위 배정되었습니다. 13명의 환자가 질병 진행, 사망, 환자 요청, 연구자의 연구약 중단 결정 또는 견딜 수 없는 독성이 발생할 때까지 erlotinib 85 mg/m2/day을 경구 투여받았습니다. Etoposide에 무작위 배정된 4명의 환자도 질병 진행 후 erlotinib을 투여받았습니다. 이 시험은 유효성 부족으로 조기 종료되었습니다. 이들 erlotinib 투여 환자 17명에서 관찰된 객관적 반응은 없었습니다.
소아 집단에서 새로운 이상사례는 확인되지 않았습니다.
암을 가진 소아 환자 105명(2~21세)을 대상으로 실시한 집단 약동학 분석에 근거할 때, CL/F/BSA(체표면적으로 표준화한 겉보기 청소율)의 기하 평균 추정치는 세 연령군에서 유사했습니다: 2~6세(n = 29), 7~16세(n = 59), 17~21세(n = 17).
8.5 노인에서의 사용
NSCLC 및 췌장암 치료를 위한 erlotinib의 임상 연구에 참여한 1297명의 피험자 중 40%가 65세 이상이었고 10%는 75세 이상이었습니다. 65세 이상과 65세 미만 피험자 간에 전반적인 안전성이나 유효성의 차이는 관찰되지 않았습니다.
8.6 간장애
간기능이 정상인 환자에서 erlotinib 치료 시 치명적인 경우를 포함한 간부전 및 간신증후군이 발생할 수 있습니다. 기저 간장애 환자에서 간독성의 위험이 증가합니다[경고 및 주의사항(5.3), 이상반응(6.1, 6.2), 용법·용량 참조]. 간장애 환자(총 빌리루빈이 정상 상한치(ULN)를 초과하거나 Child-Pugh A, B 및 C)에서 erlotinib 치료 중 모니터링하십시오. 총 빌리루빈이 3 x ULN을 초과하는 환자에서 증가된 모니터링과 함께 erlotinib 치료를 사용해야 합니다[경고 및 주의사항(5.3), 이상반응(6.1, 6.2), 용법·용량(2.4) 참조].
10 과다 복용
과다 복용 또는 과다 복용이 의심되는 환자의 경우 erlotinib 투여를 중단하고 대증 치료를 시작하십시오.
11 설명
Kinase 억제제인 Erlotinib은 화학명이 N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine인 quinazolinamine입니다. Erlotinib 정제는 다음과 같은 구조식을 가진 hydrochloride 염으로서 erlotinib을 함유합니다:
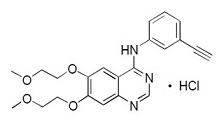
Erlotinib hydrochloride의 분자식은 C22H23N3O4.HCl이며 분자량은 429.9입니다. 이 분자는 25°C에서 pKa가 5.42입니다. 이는 formic acid에 자유롭게 용해되고, N,N-Dimethyl formamide에는 매우 약간 용해되며, 물에는 거의 용해되지 않습니다.
경구 투여용 Erlotinib 정제는 erlotinib hydrochloride (27.3 mg, 109.3 mg 및 163.9 mg) 및 다음과 같은 비활성 성분을 포함하는 25 mg, 100 mg 및 150 mg의 erlotinib 등 세 가지 용량으로 제공됩니다: lactose monohydrate, microcrystalline cellulose, sodium starch glycolate, magnesium stearate, sodium lauryl sulfate. 필름 코팅 비활성 성분으로는 hypromellose, hydroxypropyl cellulose, titanium dioxide, polyethylene glycol 400이 있습니다.
12 임상약리
12.1 작용기전
표피성장인자수용체(EGFR)는 정상세포와 암세포 모두의 세포 표면에 발현된다. 일부 종양세포에서는 EGFR 돌연변이 상태와 무관하게 이 수용체를 통한 신호전달이 종양세포의 생존과 증식에 중요한 역할을 한다. Erlotinib은 EGFR의 kinase 활성을 가역적으로 억제하여, 수용체와 관련된 tyrosine 잔기의 autophosphorylation을 방지함으로써 하위 신호전달을 억제한다. Erlotinib의 결합 친화력은 EGFR exon 19 삭제 또는 exon 21 (L858R) 돌연변이에 대해 wild type 수용체보다 높다. 다른 tyrosine kinase 수용체에 대한 Erlotinib의 억제 작용은 아직 완전히 규명되지 않았다.
12.3 약물동태학
흡수
Erlotinib은 경구 투여 후 약 60% 흡수된다. 최고 혈장 농도는 투약 4시간 후에 도달한다.
음식의 영향
음식은 erlotinib의 생체이용률을 약 100%까지 증가시켰다.
분포
Erlotinib은 혈장 albumin 및 alpha-1 acid glycoprotein (AAG)에 93% 단백결합한다.
Erlotinib의 분포용적은 232 L이다.
제거
Erlotinib은 단독요법으로 2/3차 치료를 받은 환자에서 중앙값 36.2시간의 반감기로 제거된다. 따라서 정상 상태 혈장 농도에 도달하는 시간은 7~8일이 될 것이다.
대사
Erlotinib은 in vitro에서 주로 CYP3A4에 의해, 그리고 더 적은 정도로 CYP1A2와 간외 isoform인 CYP1A1에 의해 대사된다.
배설
100 mg 경구 투여 후, 투여량의 91%가 회수되었다. 83%는 대변으로(투여량의 1%는 미변화체), 8%는 소변으로(투여량의 0.3%는 미변화체) 배설되었다.
특정 집단
2/3차 치료 또는 유지요법으로 erlotinib 단독요법을 받은 NSCLC 환자와 gemcitabine과 병용 투여한 췌장암 환자에서 연령, 체중, 성별 모두 erlotinib의 전신 노출에 임상적으로 유의한 영향을 미치지 않았다. 신장 기능이 저하된 환자에서의 erlotinib 약물동태학은 알려져 있지 않다.
간 기능 저하 환자
In vitro 및 in vivo 증거는 erlotinib이 주로 간에서 제거됨을 시사한다. 그러나 중등도의 간 기능 장애 환자(Child-Pugh B)에서의 erlotinib 노출은 원발성 간암 또는 간 전이를 포함한 적절한 간 기능을 가진 환자와 유사했다.
흡연자
건강한 지원자를 대상으로 한 단회 투여 약물동태 시험에서, 흡연(중등도 CYP1A2 inducer)은 이전/비흡연자에 비해 현재 흡연자의 erlotinib 청소율을 증가시키고 AUC0-inf를 64% (95% CI, 46~76%) 감소시켰다. NSCLC 임상시험에서, 현재 흡연자는 이전 흡연자 또는 비흡연 환자에 비해 약 2배 낮은 erlotinib 정상 상태 최저 혈장 농도에 도달했다. 이 효과는 erlotinib의 겉보기 혈장 청소율이 24% 증가한 것과 관련이 있었다. 현재 흡연자인 NSCLC 환자를 대상으로 수행된 또 다른 연구에서, 정상 상태에서의 약물동태 분석 결과 erlotinib 용량을 150 mg에서 300 mg으로 증량했을 때 용량에 비례하여 노출이 증가하는 것으로 나타났다. [용법용량 (2.4), 약물상호작용 (7), 환자상담정보 (17) 참조].
약물상호작용 연구
Gemcitabine 병용 투여는 erlotinib의 혈장 청소율에 영향을 미치지 않았다.
CYP3A4 억제제
강력한 CYP3A4 억제제인 ketoconazole과 병용 투여 시 erlotinib AUC가 67% 증가했다. CYP3A4와 CYP1A2 억제제를 병용한 ciprofloxacin은 erlotinib 노출[AUC]을 39% 증가시켰고, 최고 농도[Cmax]를 17% 증가시켰다. [용량 조절 (2.4), 약물상호작용 (7) 참조].
CYP3A4 유도제
Erlotinib 투여 7~11일 전에 CYP3A4 유도제인 rifampicin으로 전처치하면 erlotinib AUC가 58~80% 감소했다 [용량 조절 (2.4), 약물상호작용 (7) 참조].
CYP1A2 유도제 또는 흡연
특정 집단 섹션 참조 [용량 조절 (2.4), 약물상호작용 (7) 참조].
위 pH를 증가시키는 약물
Erlotinib의 용해도는 pH에 의존적이며 pH가 증가함에 따라 감소한다. Proton pump inhibitor (omeprazole)를 erlotinib과 병용 투여했을 때, erlotinib 노출[AUC]은 46% 감소했고, 최고 농도[Cmax]는 61% 감소했다. Erlotinib을 H-2 수용체 길항제(ranitidine) 300 mg 투여 2시간 후에 투여했을 때, erlotinib AUC는 33% 감소했고, Cmax는 54% 감소했다. Erlotinib을 ranitidine 150 mg 1일 2회(이전 저녁 ranitidine 투여 후 최소 10시간 후, 아침 ranitidine 투여 2시간 전)와 함께 투여했을 때, erlotinib AUC는 15% 감소했고, Cmax는 17% 감소했다 [용량 조절 (2.4), 약물상호작용 (7) 참조].
13 비임상 독성학
13.1 발암성, 변이원성, 생식능력 장애
마우스와 랫드를 대상으로 erlotinib 경구 투여량 최대 60 mg/kg/day (마우스), 5 mg/kg/day (암컷 랫드), 10 mg/kg/day (수컷 랫드)로 2년간 발암성 연구를 수행하였다. 이 연구에서 발암성 소견은 발견되지 않았다. 마우스에서 시험한 최고 용량에서의 노출량은 사람에서 erlotinib 150 mg/day 투여 시의 노출량의 약 10배였다. 수컷 랫드에서 평가한 최고 용량에서의 노출량은 사람에서의 노출량의 2배였고, 암컷 랫드에서 시험한 최고 용량에서의 노출량은 사람에서의 노출량보다 약간 낮았다.
Erlotinib은 일련의 in vitro 시험(박테리아 돌연변이, 인체 림프구 염색체 이상 및 포유류 세포 돌연변이)과 in vivo 마우스 골수 소핵 시험에서 유전적 손상을 일으키지 않았다.
Erlotinib은 수컷 또는 암컷 랫드의 생식능력을 손상시키지 않았다.
14 임상 연구
14.1 비소세포폐암(NSCLC) – EGFR 변이를 가진 환자의 첫 번째 치료
연구 1
EGFR exon 19 결손 또는 exon 21 (L858R) 치환 돌연변이를 가진 전이성 NSCLC 환자에 대한 엘로티닙의 단독요법 일차 치료의 안전성과 효능은 유럽에서 실시된 무작위 배정, 공개 임상시험인 연구 1에서 입증되었습니다. 174명의 백인 환자가 엘로티닙 150mg을 1일 1회 질병 진행 시까지 투여(n=86) 또는 표준 백금 기반 복합 화학요법(n=88) 4주기 투여에 1:1로 무작위 배정되었습니다. 표준 화학요법 요법은 cisplatin과 gemcitabine, cisplatin과 docetaxel, carboplatin과 gemcitabine, carboplatin과 docetaxel 병용요법이었습니다. 주요 유효성 평가변수는 연구자가 평가한 무진행 생존기간(PFS)이었습니다. 무작위 배정은 EGFR 변이(exon 19 결손 또는 exon 21 (L858R) 치환)와 Eastern Cooperative Oncology Group Performance Status (ECOG PS)(0 vs. 1 vs. 2)에 따라 층화되었습니다. 환자 선별 및 등록을 위한 EGFR 변이 상태는 임상시험용 검사법(CTA)으로 결정되었습니다. 엘로티닙 투여군 69명 및 화학요법군 65명 등 총 134명 환자의 종양 검체가 FDA 승인 동반진단 cobas® EGFR 돌연변이 검사로 사후 검사되었습니다.
전체 연구 인구집단의 기준 인구통계학적 특성은 다음과 같습니다: 여성(72%), 백인(99%), 연령 ≥65세(51%), ECOG PS 1(53%), ECOG PS 0(33%), ECOG PS 2(14%), 현재 흡연자(11%), 과거 흡연자(20%), 비흡연자(69%). 질병 특성으로는 제6판 AJCC(American Joint Commission on Cancer) 분류에 따라 병기 IV가 93%, 병기 IIIb 늑막삼출이 7%, 93%가 adenocarcinoma, CTA에 따라 66%가 exon 19 결손, 34%가 exon 21 (L858R) 돌연변이가 있었습니다.
연구자 평가에 따른 PFS(RECIST 1 또는 임상적 진행 기준)에서 엘로티닙 투여군이 화학요법군에 비해 통계적으로 유의한 개선 효과를 보였습니다(표 6 및 그림 1 참조). 독립적 평가위원회가 평가한 환자군(연구 1 대상자의 약 75%)과 cobas® EGFR 돌연변이 검사로 EGFR 변이가 확인된 134명(연구 1 인구집단의 77%)에서도 유사한 PFS(RECIST 1 기준) 결과를 보였습니다.
PFS의 최종 분석 시점에 실시된 프로토콜 규정 전체 생존기간(OS) 분석에서는 엘로티닙군과 화학요법군 간에 통계적으로 유의한 차이가 없었습니다. 자료 절단 시점에 화학요법군의 84%가 최소 1회 이상 후속 치료를 받았고, 이 중 97%가 EGFR-tyrosine kinase 억제제를 투여받았습니다. 엘로티닙군에서는 66%가 최소 1회 이상 후속 치료를 받았습니다.
표 6: 유효성 결과(연구 1)
| 유효성 평가변수 |
엘로티닙 (N = 86) |
화학요법 (N = 88) |
|---|---|---|
| 무진행 생존기간 |
||
| 진행 또는 사망 환자 수 | 71명(83%) | 63명(72%) |
| 중앙값 PFS(개월) (95% CI) | 10.4 (8.7, 12.9) | 5.2 (4.6, 6) |
| 위험비 (95% CI)(1) |
0.34 (0.23, 0.49) | |
| p-값 (비층화 로그순위 검정) | < 0.001 | |
| 전체 생존기간 |
||
| 사망 환자 수(%) | 55명(64%) | 54명(61%) |
| 중앙값 OS(개월) (95% CI) | 22.9 (17, 26.8) | 19.5 (17.3, 28.4) |
| 위험비 (95% CI)1 |
0.93 (0.64, 1.35) | |
| 객관적 반응률 |
||
| 객관적 반응률 (95% CI) | 65% (54.1%, 75.1%) | 16% (9%, 25.3%) |
(1) 비층화 Cox 회귀모델.
그림 1: 연구 1의 연구자 평가 PFS Kaplan-Meier 곡선
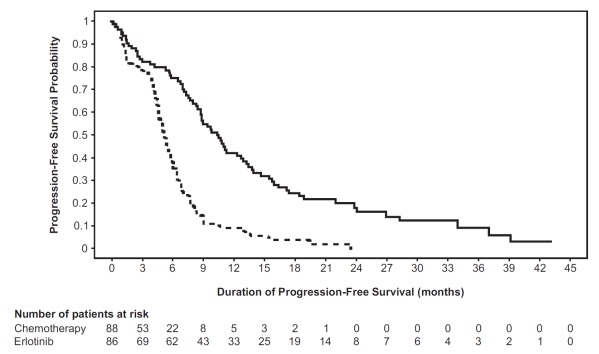
EGFR 변이 아형에 따른 탐색적 하위그룹 분석에서 PFS에 대한 위험비(HR)는 exon 19 결손 환자군에서 0.27(95% CI 0.17-0.43), exon 21 (L858R) 치환 환자군에서 0.52(95% CI 0.29-0.95)였습니다. OS에 대한 위험비는 exon 19 결손 하위그룹에서 0.94(95% CI 0.57-1.54), exon 21 (L858R) 치환 하위그룹에서 0.99(95% CI 0.56-1.76)였습니다.
14.2 NSCLC – EGFR 변이가 없는 환자의 유지 요법으로서 Erlotinib의 효과 부재
EGFR 활성화 변이가 없는 NSCLC 환자에서 유지 요법으로 Erlotinib의 효과가 없음이 연구 2에서 입증되었습니다. 연구 2는 EGFR exon 19 또는 exon 21 L858R 돌연변이가 없는 진행성 NSCLC 환자 643명을 대상으로 한 다기관, 위약 대조군, 무작위 배정 임상시험이었습니다. 4주기의 백금 기반 화학요법 후에도 질병이 진행하지 않은 환자들을 1:1로 나누어 Erlotinib 150mg 또는 위약을 경구 1일 1회 투여하였습니다(Erlotinib 322명, 위약 321명). 초기 요법 진행 후에는 공개 라벨 단계로 넘어갈 수 있었습니다. 기저 특성은 다음과 같았습니다: 중앙값 연령 61세(65세 이상 35%), 남성 75%, 백인 77%, 아시아인 21%, ECOG PS 0 28%, ECOG PS 1 72%, 비흡연자 16%, 현재 흡연자 58%, 선암 57%, 편평세포암 35%, 복합 모드 치료에 적합하지 않은 병기 IIIb 22%, 병기 IV 78%. Erlotinib 군에서 50%, 위약군에서 77%가 공개 라벨 단계로 진행하여 화학요법 또는 Erlotinib를 받았습니다.
주요 평가변수는 전체 생존기간(OS)이었습니다. 중앙 OS는 Erlotinib군 9.7개월, 위약군 9.5개월이었으며, 위험비는 1.02(95% CI 0.85, 1.22)였습니다. 중앙 무진행생존기간(PFS)은 Erlotinib군 3개월, 위약군 2.8개월이었으며, 위험비는 0.94(95% CI 0.8, 1.11)였습니다.
14.3 NSCLC – 유지 요법 또는 2차/3차 치료
무작위 배정, 이중 맹검, 위약 대조 임상시험 연구 3과 연구 4에서 화학요법 초기 치료 후 유지 요법으로(연구 3) 또는 화학요법 초기 치료 후 병력 진행 시(연구 4) 전이성 NSCLC 환자에게 Erlotinib를 투여한 효능 및 안전성을 평가하였습니다. 이 연구들에서는 EGFR 돌연변이 여부와 무관하게 등록되었습니다.
연구 3
NSCLC의 유지 요법 치료제로서 Erlotinib의 효능과 안전성이 연구 3에서 입증되었습니다. 연구 3은 1차 백금 기반 화학요법 중 질병이 진행하지 않은 전이성 NSCLC 환자 889명을 대상으로 한 무작위 배정, 이중 맹검, 위약 대조 임상시험으로, 26개국에서 수행되었습니다. 환자들을 1:1로 나누어 Erlotinib 150mg 또는 위약을 경구 1일 1회 투여하였습니다(Erlotinib 438명, 위약 451명). 이 연구의 주요 목적은 NSCLC 치료에서 표준 백금 기반 화학요법 후 Erlotinib 투여가 전체 환자군 또는 EGFR 면역조직화학염색(IHC) 양성 종양 환자군에서 위약과 비교하여 무진행생존기간(PFS) 향상을 가져오는지 확인하는 것이었습니다.
전체 연구 대상군의 기저 인구통계학적 특성은 다음과 같습니다: 남성(74%), 65세 미만(66%), ECOG PS 1(69%), ECOG PS 0(31%), 백인(84%), 아시아인(15%), 현재 흡연자(55%), 과거 흡연자(27%), 비흡연자(17%). 질병 특성은 다음과 같습니다: 병기 IV(75%), 삼출액을 동반한 병기 IIIb(25%)(AJCC 6판 기준), 선암(기관지폐포암 포함) 45%, 편평세포암 40%, 대세포암 5%; EGFR IHC 양성 70%, 음성 14%, 불확정 4%, 미상 12%.
표 7: 유효성 결과(연구 3): (ITT 집단)1
| 유효성 평가변수 |
Erlotinib (N = 438) |
위약 (N = 451) |
| 연구자 평가에 따른 무진행생존기간(PFS) |
||
| 진행 또는 사망 환자 수(%) | 349 (80%) | 400 (89%) |
| 중앙 PFS (개월)(95% CI) | 2.8 (2.8, 3.1) | 2.6 (1.9, 2.7) |
| 위험비 (95% CI)(2) |
0.71 (0.62, 0.82) | |
| p-값(층화 로그순위 검정) (2,3) |
p < 0.0001 | |
| 전체 생존기간(OS) |
||
| 사망자 수 | 298 (68%) | 350 (78%) |
| 중앙 OS(개월)(95% CI) | 12 (10.6, 13.9) | 11 (9.9, 12.1) |
| 위험비(95% CI) (2) |
0.81 (0.7, 0.95) | |
| p-값(층화 로그순위 검정) (3) |
0.0088 | |
(1) 무작위 배정 전 진행을 보인 환자는 PFS 및 TTP 분석에서 제외되었습니다.
(2) 단변량 Cox 회귀모형
(3) 비층화 로그순위 검정
그림 2는 연구 3에서 치료군별 전체 생존기간에 대한 Kaplan-Meier 곡선을 나타냅니다.
그림 2: 연구 3의 치료군별 전체 생존기간에 대한 Kaplan-Meier 곡선
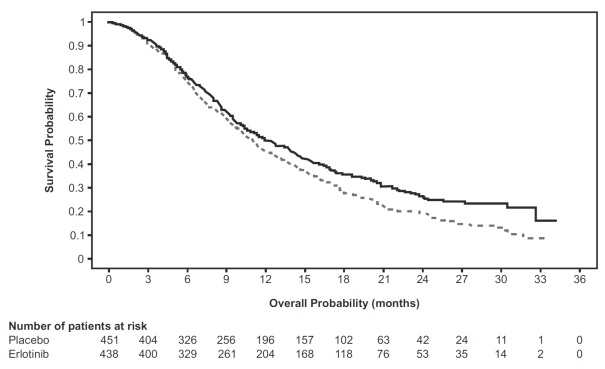
주: 위험비(HR)는 단변량 Cox 회귀모형에서 산출되었습니다.
연구 4
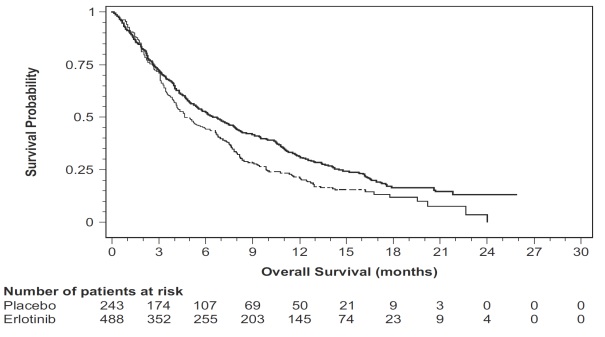
14 임상 연구
단일제 erlotinib의 유효성과 안전성은 Study 4에서 평가되었다. 이는 최소 한 가지 화학요법 치료 실패 후 국소 진행성 또는 전이성 NSCLC 환자 731명을 대상으로 한 무작위, 이중 맹검, 위약 대조 시험이었다. 환자들은 2:1의 비율로 경구 erlotinib 150 mg 또는 위약(erlotinib 488명, 위약 243명)을 1일 1회 복용하도록 무작위 배정되었으며, 질병 진행 또는 허용할 수 없는 독성이 나타날 때까지 계속하였다. 유효성 평가 항목에는 전체 생존기간, 반응률, 무진행 생존기간(PFS)이 포함되었다. 반응 지속기간도 평가되었다. 1차 평가변수는 생존률이었다. 이 연구는 17개국에서 수행되었다.
전체 연구 대상 환자군의 기준선 인구통계학적 특성은 다음과 같다: 남성(65%), 백인(78%), 아시아인(12%), 흑인(4%), 65세 미만(62%), ECOG PS 1(53%), ECOG PS 0(13%), ECOG PS 2(25%), ECOG PS 3(9%), 현재 또는 과거 흡연자(75%), 비흡연자(20%), 이전 백금 기반 요법 경험(93%). 종양 특성은 다음과 같다: 선암(50%), 편평상피암(30%), 미분화 대세포암(9%), 혼합 비소세포암(2%).
연구 결과는 표 8에 나와 있다.
표 8: 유효성 결과(Study 4)
| 유효성 평가변수 | Erlotinib (N = 488) |
위약 (N = 243) |
| 전체 생존율(OS) | ||
| 사망자 수 | 378명(77%) | 209명(86%) |
| 중앙값 OS(개월)(95% CI) | 6.7 (5.5, 7.8) | 4.7 (4.1, 6.3) |
| 위험비(95% CI) (1) | 0.73 (0.61, 0.86) | |
| p-값(층화 로그순위 검정) (2) | p < 0.001 | |
| 무진행 생존기간(PFS) | ||
| 진행 또는 사망 건수(%) | 402건(82%) | 211건(87%) |
| 중앙값 PFS(개월)(95% CI) | 2.3 (1.9, 3.3) | 1.8 (1.8, 1.9) |
| 위험비(95% CI) 1 | 0.59 (0.5, 0.7) | |
| 객관적 반응 | ||
| 객관적 반응률(95% CI) | 8.9% (6.4, 12) | 0.9% (0.1, 3.4) |
(1) ECOG 수행 상태, 이전 요법 수, 이전 백금 치료, 이전 화학요법에 대한 최상의 반응을 공변량으로 한 Cox 회귀모델
(2) ECOG 수행 상태, 이전 요법 수, 이전 백금 치료, 이전 화학요법에 대한 최상의 반응을 층화변수로 한 양측 로그순위 검정
그림 3은 전체 생존기간에 대한 Kaplan-Meier 곡선을 나타낸다.
그림 3: Study 4에서 치료군별 전체 생존기간에 대한 Kaplan-Meier 곡선
14.4 NSCLC – 화학요법과 병용 투여된 Erlotinib의 무효능
국소 진행성 또는 전이성 NSCLC 1차 치료 환자를 대상으로 한 두 건의 다기관, 위약 대조, 무작위 배정 시험(erlotinib군, N=526명 또는 N=580명)에서 백금 기반 화학요법[carboplatin 및 paclitaxel 또는 gemcitabine 및 cisplatin]과 병행하여 erlotinib를 투여했을 때 임상적 유익성이 관찰되지 않았다.
14.5 췌장암 – Gemcitabine과 병용 투여된 Erlotinib
1차 치료제로서 gemcitabine과 병용 투여된 erlotinib의 유효성과 안전성은 Study 5에서 평가되었다. 이는 국소 진행성, 수술 불가능 또는 전이성 췌장암 환자 569명을 대상으로 한 무작위, 이중 맹검, 위약 대조 시험이었다. 환자들은 1:1의 비율로 erlotinib(100 mg 또는 150 mg) 또는 위약을 1일 1회 연속 복용하고, gemcitabine을 정맥 주사(1000 mg/m2, Cycle 1 – 1, 8, 15, 22, 29, 36 및 43일, Cycle 2 이후 – 4주 주기의 1, 8, 15일[췌장암에 대한 승인 용량 및 투여 간격, gemcitabine 제품설명서 참조])로 투여받도록 무작위 배정되었다. Erlotinib 또는 위약은 질병 진행 또는 허용할 수 없는 독성 증상이 나타날 때까지 경구 1일 1회 복용하였다. 1차 평가변수는 생존률이었다. 2차 평가변수에는 반응률, 무진행 생존기간(PFS)이 포함되었다. 반응 지속기간도 평가되었다. 이 연구는 18개국에서 수행되었다. Gemcitabine과 erlotinib을 병용 투여받은 환자는 총 285명(100 mg군 261명, 150 mg군 24명)이었고, gemcitabine과 위약을 투여받은 환자는 총 284명(100 mg군 260명, 150 mg군 24명)이었다. 150 mg군은 환자 수가 너무 적어 결론을 내리기 어려웠다.
100 mg군에서 전체 연구 대상 환자군의 기준선 인구통계학적 특성은 다음과 같다: 남성(52%), 백인(88%), 아시아인(7%), 흑인(2%), 65세 미만(53%), ECOG PS 1(51%), ECOG PS 0(32%), ECOG PS 2(17%). Erlotinib군에서 여성 비율이 위약군에 비해 약간 더 높았다(51% vs. 44%). 초기 진단부터 무작위 배정까지의 중앙 기간은 약 1개월이었다. 대부분의 환자(76%)는 기준선에서 원격 전이가 있었고, 24%는 국소 진행성 병변이 있었다.
연구 결과는 표 9에 나와 있다.
표 9: 유효성 결과: Erlotinib 100 mg군(Study 5)
| 효능 매개변수 |
Erlotinib + Gemcitabine (N = 261) |
Placebo + Gemcitabine (N = 260) |
|---|---|---|
| 전체 생존율(OS) |
||
| 사망 수 | 250 | 254 |
| 중앙값 OS 개월(95% CI) | 6.5 (6, 7.4) | 6 (5.1, 6.7) |
| 위험비(95% CI)(1) |
0.81 (0.68, 0.97) | |
| p-값 (계층화된 로그-순위 검정)(2) |
0.028 | |
| 무진행 생존율(PFS) |
||
| 진행 또는 사망 수(%) | 225 | 232 |
| 중앙값 PFS 개월(95% CI) | 3.8 (3.6, 4.9) | 3.6 (3.3, 3.8) |
| 위험비(95% CI) (1) |
0.76 (0.64, 0.92) | |
| 객관적 반응률 |
||
| 객관적 반응률(95% CI) | 8.6% (5.4, 12.9) | 7.9% (4.8, 12) |
(1) ECOG 활동능력 등급과 질병 범위 공변량을 포함한 Cox 회귀 모델.
(2) ECOG 활동능력 등급과 질병 범위를 계층화한 양측 로그-순위 검정.
생존율은 의도 치료 대상 인구집단에서 평가되었다. 그림 4는 100mg 집단에서의 전체 생존율에 대한 Kaplan-Meier 곡선을 나타낸다. 주요 생존율 및 PFS 분석은 ECOG 활동능력 등급과 질병 범위를 계층화한 양측 로그-순위 검정이었다.
그림 4: 연구 5에서 100mg 집단의 전체 생존율에 대한 Kaplan-Meier 곡선
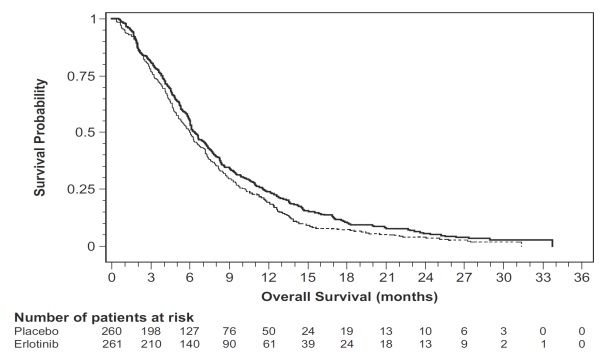
참고: HR은 ECOG 활동능력 등급과 질병 범위 공변량을 포함한 Cox 회귀 모델에서 산출되었다. p-값은 ECOG 활동능력 등급과 질병 범위를 계층화한 양측 로그-순위 검정에서 산출되었다.
16 제공/보관 및 취급 방법
Erlotinib 정제 25 mg은 한쪽 면에 “L55″가 음각되어 있고 다른 쪽 면은 평평한 둥근 쌍볼록 흰색 필름코팅정입니다.
어린이 안전 잠금장치가 있는 30정 들이 병, NDC 62332-565-30
어린이 안전 잠금장치가 있는 90정 들이 병, NDC 62332-565-90
Erlotinib 정제 100 mg은 한쪽 면에 “L630″이 음각되어 있고 다른 쪽 면은 평평한 둥근 쌍볼록 흰색 필름코팅정입니다.
어린이 안전 잠금장치가 있는 30정 들이 병, NDC 62332-566-30
어린이 안전 잠금장치가 있는 90정 들이 병, NDC 62332-566-90
Erlotinib 정제 150 mg은 한쪽 면에 “L631″이 음각되어 있고 다른 쪽 면은 평평한 둥근 쌍볼록 흰색 필름코팅정입니다.
어린이 안전 잠금장치가 있는 30정 들이 병, NDC 62332-567-30
어린이 안전 잠금장치가 있는 90정 들이 병, NDC 62332-567-90
25°C(77°F)에서 보관하며, 15°~30°C(59°~86°F)의 편차가 허용됩니다. USP Controlled Room Temperature 참조.
17 환자 상담 정보
피부 발진, 수포성 및 박리성 피부 장애
- erlotinib 정제를 복용하는 동안 햇빛에 노출된 부위에서 피부 반응이 발생하거나 악화될 수 있음을 환자에게 알리고, 알코올이 포함되지 않은 연화 크림 및 자외선 차단제 사용 또는 햇빛 노출 피하기와 같은 사전 조치가 포함될 수 있습니다. 과색소침착 또는 건조한 피부가 digital skin fissures를 동반하거나 동반하지 않고 보고되었으며, 대부분의 경우 발진과 관련이 있음을 환자에게 알립니다[이상반응(6.1) 참조].
- erlotinib 정제가 수포성 및 박리성 피부 장애의 위험을 증가시킬 수 있으며, 중증 피부 반응 시 즉시 의학적 주의를 구할 것을 환자에게 알립니다[경고 및 주의사항(5.5) 참조].
설사
설사는 보통 loperamide로 조절할 수 있으며, 중증 또는 지속적인 설사의 경우 의료 서비스 제공자에게 연락하도록 환자에게 조언합니다[이상반응(6.1) 참조].
간질성 폐질환
폐렴을 포함한 중증 또는 치명적인 ILD의 위험에 대해 환자에게 알립니다. 새로운 또는 악화되는 원인 불명의 호흡 곤란 또는 기침을 즉시 보고하도록 환자에게 조언합니다[용량 및 투여(2.4) 및 경고와 주의사항(5.1) 참조].
신부전
신부전 발생의 위험에 대해 환자에게 알립니다. 의료 서비스 제공자가 신장 기능 및 전해질을 모니터링해야 할 필요성에 대해 환자에게 알립니다[경고 및 주의사항(5.2) 참조].
간독성
간독성의 징후 또는 증상을 즉시 보고하도록 환자에게 조언합니다[경고 및 주의사항(5.3) 참조].
위장관 천공
erlotinib 정제가 위장관 천공 또는 누공의 위험을 증가시킬 수 있으며, 중증 복통 시 즉시 의학적 주의를 구할 것을 환자에게 조언합니다[용량 및 투여(2.4) 및 경고와 주의사항(5.4) 참조].
뇌혈관 사고
뇌혈관 사고의 위험성에 대해 환자에게 알리고 즉시 의학적 주의를 구하도록 합니다[용량 및 투여(2.4) 및 경고와 주의사항(5.6) 참조].
안과적 장애
유루증, 광과민성, 시야 흐림, 안구 통증, 충혈 또는 시력 변화와 같은 안구 증상이나 징후가 발생하면 즉시 의료 서비스 제공자에게 연락하도록 환자에게 조언합니다[용법 용량(2.4) 및 경고와 주의사항(5.8) 참조].
와파린 복용 환자의 출혈
와파린을 투여 받는 환자에게 INR 또는 기타 coumarin 유도체 항응고제를 모니터링해야 할 필요성에 대해 조언합니다[경고 및 주의사항(5.9) 및 약물 상호작용(7) 참조].
모발 및 손발톱 장애
다모증, 손발톱 부서짐 및 손발톱 헐거워짐 등 모발 및 손발톱 장애가 보고되었음을 환자에게 조언합니다[이상반응(6.1) 참조].
태아 독성
- 임신부 및 가임기 여성에게 태아에 대한 잠재적 위험에 대해 알립니다. 가임기 여성은 알려진 또는 의심되는 임신 사실을 의료 서비스 제공자에게 알리도록 권고합니다[경고 및 주의사항(5.10), 특정 집단에서의 사용(8.1) 참조].
- 가임기 여성에게 erlotinib 정제로 치료하는 동안 그리고 마지막 투여 후 1개월 동안 효과적인 피임법을 사용하도록 조언합니다[특정 집단에서의 사용(8.3) 참조].
수유
- erlotinib 정제로 치료하는 동안 그리고 최종 투여 후 2주 동안 모유 수유를 하지 않도록 여성에게 조언합니다[특정 집단에서의 사용(8.2) 참조].
흡연
- 흡연 상태에 변화가 있는 경우 의료 서비스 제공자에게 연락하고 흡연하는 경우 erlotinib 정제의 용량을 조정해야 할 수 있음을 환자에게 조언합니다[약물 상호 작용(7) 및 임상 약리학(12.3) 참조]
- 금연하도록 환자에게 조언합니다[임상 약리학(12.3) 참조].
부작용에 대한 의학적 조언은 의사에게 문의하십시오. 1-800-FDA-1088로 FDA에 부작용을 보고할 수 있습니다.
자세한 내용은 1-866-210-9797로 전화하십시오.
나열된 브랜드는 해당 소유자의 상표입니다.
제조사:
Alembic Pharmaceuticals Limited
Formulation Division II, Survey No. 84,
87 And 88, Panelav, Taluka Halol,
Panchmahal, Gujarat 389350, India
판매사:
Alembic Pharmaceuticals, Inc.
Bedminster, NJ 07921, USA
개정일: 02/2023
패키지 라벨.주요 표시면 25 mg
NDC 62332-565-30
Erlotinib 정제
25 mg
전문의약품
30 정
Alembic

패키지 라벨.주요 표시부 100 mg
NDC 62332-566-30
Erlotinib Tablets
100 mg
Rx only
30 정
Alembic

포장 라벨.주요 표시 패널 150 mg
NDC 62332-567-30
Erlotinib 정제
150 mg
처방전 필요
30 정
Alembic