
의약품 제조업체: Genentech, Inc. (Updated: 2024-04-30)
사용 설명서 하이라이트
ALECENSA® (alectinib) 캡슐, 경구 투여
최초 미국 승인: 2015년
적응증
투여 및 투여량
경구 1일 2회 600mg. ALECENSA를 음식과 함께 복용하십시오. (2.2)
제형 및 강도
캡슐: 150mg (3)
금기 사항
없음. (4)
경고 및 주의사항
- 간독성: 치료 첫 3개월 동안 2주마다, 그 후에는 한 달에 한 번씩, 그리고 임상적으로 필요한 경우 더 자주 간 기능 검사를 실시하고 트랜스아미나제와 빌리루빈 수치가 상승한 환자에서는 더 자주 검사를 실시하십시오. ALT, AST 또는 빌리루빈 수치가 심각하게 상승하는 경우 ALECENSA 투여를 중단한 후 감량하거나 영구적으로 투여를 중단하십시오. (2.4, 5.1)
- 간질성폐질환(ILD)/폐렴: ILD/폐렴으로 진단된 환자에서는 즉시 ALECENSA 투여를 중단하고, 다른 잠재적 원인이 확인되지 않으면 영구적으로 중단하십시오. (2.4, 5.2)
- 신장애: 중증 신장애가 있는 경우 ALECENSA 투여를 중단한 후 회복 시 감량하여 재투여하거나 영구적으로 투여를 중단하십시오(2.4, 5.3).
- 서맥: 심박수와 혈압을 정기적으로 모니터링하십시오. 증상이 있는 경우 ALECENSA 투여를 중단한 후 감량하거나 영구적으로 투여를 중단하십시오. (2.4, 5.4)
- 심각한 근육통 및 크레아틴 인산 활성 효소(CPK) 상승: 치료 첫 달 동안 2주마다, 그리고 설명할 수 없는 근육통, 압통 또는 근력 약화를 보이는 환자에서 CPK 수치를 평가하십시오. CPK 수치가 심각하게 상승하는 경우 투여를 중단한 후 재개하거나 감량하십시오. (2.4, 5.5)
- 용혈성 빈혈: 만약 용혈성 빈혈이 의심되면 ALECENSA 투여를 중단하십시오. 용혈성 빈혈이 확인되면 회복 시 감량하여 재투여하거나 영구적으로 투여를 중단하는 것을 고려하십시오. (5.6)
- 태아 독성: ALECENSA는 태아에 해를 끼칠 수 있습니다. 가임 여성에게 태아에 대한 잠재적 위험과 효과적인 피임법 사용에 대해 조언하십시오. (5.7, 8.1, 8.3)
이상반응
가장 흔한 이상반응(발생률 ≥20%)은 간독성, 변비, 피로, 근육통, 부종, 발진 및 기침이었습니다. (6.1)
의심되는 이상반응이 있는 경우 Genentech(1-888-835-2555) 또는 FDA(1-800-FDA-1088 또는 www.fda.gov/medwatch)에 연락하십시오.
환자 상담 정보 및 FDA 승인 환자 용기 라벨 정보는 17을 참조하십시오.
개정: 4/2024
목차
전체 처방 정보: 내용*
1 적응증 및 사용법
1.1 절제된 ALK 양성 비소세포폐암 (NSCLC)의 보조 치료
1.2 전이성 ALK 양성 NSCLC 치료
2 투여 용량 및 투여 방법
2.1 환자 선별
2.2 투여 용량 및 투여 방법
2.3 간장애 환자에 대한 권장 투여량
2.4 이상반응에 대한 투여량 조정
3 제형 및 강도
4 금기사항
5 경고 및 주의사항
5.1 간독성
5.2 간질성 폐렴 (ILD)/폐렴
5.3 신장 장애
5.4 서맥
5.5 중증 근육통 및 크레아틴 인산화 효소 (CPK) 상승
5.6 용혈성 빈혈
5.7 배아-태아 독성
6 이상반응
6.1 임상시험 경험
8 특정 집단에서의 사용
8.1 임신
8.2 수유
8.3 생식능력이 있는 여성 및 남성
8.4 소아 사용
8.5 노인 사용
8.6 신장애
8.7 간장애
10 과량투여
11 설명
12 임상약리학
12.1 작용기전
12.2 약력학
12.3 약동학
13 비임상 독성
13.1 발암성, 돌연변이 유발성, 생식능력 저하
14 임상연구
14.1 절제된 ALK 양성 NSCLC의 보조 치료
14.2 전이성 ALK 양성 NSCLC 치료
16 공급/보관 및 취급 방법
17 환자 상담 정보
- *
- 전체 처방 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
1 적응증 및 용법
1.1 절제된 ALK-양성 비소세포폐암(NSCLC)의 보조 치료
ALECENSA는 FDA 승인 검사에서 확인된 anaplastic 림프종 운화효소(ALK)-양성 비소세포폐암(NSCLC)(종양 ≥ 4cm 또는 림프절 양성)의 종양 절제 후 성인 환자에서 보조 치료제로 적응증이 있습니다. [용법 및 투여량(2.1)참조]
1.2 전이성 ALK-양성 NSCLC 치료
ALECENSA는 FDA 승인 검사에서 확인된 ALK-양성 전이성 NSCLC 성인 환자 치료에 적응증이 있습니다. [용법 및 투여량(2.1)참조]
2 용량 및 투여
2.1 환자 선택
수술 가능한 종양이 있는 환자의 경우 종양 조직에서 ALK 양성 여부에 따라 ALECENSA를 비소세포폐암의 보조 치료제로 선택합니다 [사용 지침(1.1) 및 임상 연구(14.1) 참조].
전이성 비소세포폐암 치료를 위해 종양 조직 또는 혈장 검체에서 ALK 양성 여부에 따라 ALECENSA로 환자를 선택합니다 [사용 지침(1.2) 및 임상 연구(14.2) 참조]. 혈장 검체에서 ALK 재배열이 검출되지 않는 경우 가능하다면 종양 조직을 검사하십시오.
비소세포폐암에서 ALK 재배열 검출을 위한 FDA 승인 검사에 관한 정보는 http://www.fda.gov/CompanionDiagnostics에서 확인할 수 있습니다.
2.2 투여 및 투여 방법
ALECENSA의 권장 투여량 정보는 표 1에 나와 있습니다.
| 적응증 | ALECENSA 권장 투여량 | 기간 |
|---|---|---|
| 절제된 비소세포폐암의 보조 치료 | 식사와 함께 1일 2회 600mg 경구 투여 [임상 약리학(12.3) 참조] |
2년간 또는 질병 재발 또는 허용 불가능한 독성이 나타날 때까지 |
| 전이성 비소세포폐암 | 질병 진행 또는 허용 불가능한 독성이 나타날 때까지 | |
|
||
2.3 간장애 환자의 권장 투여량
중증 간장애(Child-Pugh C) 환자의 ALECENSA 권장 투여량은 1일 2회 450mg 경구 투여입니다 [특정 집단에서의 사용(8.7) 및 임상 약리학(12.3) 참조].
2.4 이상반응에 대한 투여량 조절
ALECENSA의 투여량 감소 일정은 표 2에 나와 있습니다.
| 투여량 감소 일정 | 투여량 수준 |
|---|---|
| 초기 투여량 | 1일 2회 600mg 경구 투여 |
| 첫 번째 투여량 감소 | 1일 2회 450mg 경구 투여 |
| 두 번째 투여량 감소 | 1일 2회 300mg 경구 투여 |
1일 600mg(300mg 씩 1일 2회 투여) 을 내성이 없을 경우 계속해서 투여합니다. 1일 600mg 투여를 견딜 수 없는 경우, 투여 중단합니다.
ALECENSA의 용량 조절 권고사항은 표 3을 참고하십시오.
| 조건* | ALECENSA 용량 조절 |
|---|---|
| ALT 또는 AST 상승 수치가 정상 상한치(ULN)의 5배 이상이나 총 빌리루빈 수치가 정상 상한치(ULN)의 2배 이하 | 기저치 또는 정상 상한치(ULN)의 3배 이하로 회복될 때까지 일시 중단 후 표 2의 감량 기준에 따라 복용을 재개합니다. |
| ALT 또는 AST 상승 수치가 정상 상한치(ULN)의 3배 초과이면서 총 빌리루빈 상승 수치가 정상 상한치(ULN)의 2배 초과인 경우(담즙정체나 용혈의 증거가 없는 경우) | ALECENSA 복용을 영구 중단합니다. |
| 총 빌리루빈 상승 수치가 정상 상한치(ULN)의 3배 초과 | 기저치 또는 정상 상한치(ULN)의 1.5배 이하로 회복될 때까지 일시 중단 후 표 2의 감량 기준에 따라 복용을 재개합니다. |
| 치료와 관련된 모든 등급의 간질성 폐질환(ILD)/폐렴 | ALECENSA 복용을 영구 중단합니다. |
| 3등급 신장애 | 혈중 크레아티닌 수치가 정상 상한치(ULN)의 1.5배 이하로 회복될 때까지 일시 중단 후 감량하여 복용을 재개합니다. |
| 4등급 신장애 | ALECENSA 복용을 영구 중단합니다. |
| 증상성 서맥 | 무증상 서맥 또는 심박수 60회/분 이상으로 회복될 때까지 ALECENSA 복용을 중단합니다. 병용 약물 복용이 원인이고 중단하거나 용량을 조절할 수 있는 경우, 무증상 서맥 또는 심박수 60회/분 이상으로 회복되면 이전 용량으로 ALECENSA 복용을 재개합니다. 병용 약물 복용이 원인으로 발견되지 않거나, 병용 약물 복용을 중단하지 않거나 용량을 조절하지 않는 경우, 무증상 서맥 또는 심박수 60회/분 이상으로 회복되면 표 2의 감량 기준에 따라 ALECENSA 복용을 재개합니다. |
| 생명을 위협하는 서맥† (긴급 중재가 필요한 경우) | 병용 약물 복용이 원인으로 발견되지 않는 경우 ALECENSA 복용을 영구 중단합니다. 병용 약물 복용이 원인이고 중단하거나 용량을 조절할 수 있는 경우, 무증상 서맥 또는 심박수 60회/분 이상으로 회복되면 표 2의 감량 기준에로 ALECENSA 복용을 재개하고 임상적으로 적절한 빈도로 모니터링합니다. 재발 시 ALECENSA 복용을 영구 중단합니다. |
| CPK 상승 수치가 정상 상한치(ULN)의 5배 초과 | 기저치 또는 정상 상한치(ULN)의 2.5배 이하로 회복될 때까지 일시 중단 후 같은 용량으로 복용을 재개합니다. |
| CPK 상승 수치가 정상 상한치(ULN)의 10배 초과 또는 5배 초과 CPK 상승이 두 번째로 발생한 경우 | 기저치 또는 정상 상한치(ULN)의 2.5배 이하로 회복될 때까지 일시 중단 후 표 2의 감량 기준에 따라 복용을 재개합니다. |
| 용혈성 빈혈 | 용혈성 빈혈이 의심되면 ALECENSA 복용을 중단합니다. 증상이 회복되면 감량하여 복용을 재개하거나 영구 중단합니다. |
3 투여 형태 및 강도
150mg 경질 캡슐, 흰색, 캡에 “ALE” 검정색 잉크로 인쇄되어 있고, 본체에 “150mg” 검정색 잉크로 인쇄되어 있습니다.
4 금기사항
없음.
5 경고 및 주의사항
5.1 간독성
ALECENSA로 치료받은 환자에서 약물 유발성 간 손상을 포함한 중증 간독성이 발생했습니다.
ALECENSA를 투여받은 환자의 통합 안전성 집단에서[이상반응 (6.1)을 참조하십시오] 41%에서 간독성이 발생했고, 3등급 이상 간독성의 발생률은 8%였습니다. ALINA 연구에서 ALECENSA로 치료받은 환자의 61%에서 간독성이 발생했고 3등급 이상 간독성의 발생률은 4.7%였습니다. 대부분(136명 중 72%)의 트랜스아미나제 상승은 치료 첫 3개월 동안 발생했습니다. 통합 안전성 집단에서 ALECENSA를 투여받은 환자의 3.6%에서 간독성으로 인한 치료 중단이 있었으며, ALINA 연구에서는 1.6%의 환자에서 간독성으로 인한 치료 중단이 있었습니다.
통합 안전성 집단에서 ALECENSA로 치료받은 환자의 1% 미만에서 정상 알칼리성 인산가수분해효소와 함께 ALT 또는 AST가 정상 상한치의 3배 이상, 총 빌리루빈이 정상 상한치의 2배 이상으로 동시에 상승했습니다. 3-4등급의 AST/ALT 상승이 있었던 3명의 환자에서 약물 유발성 간 손상(2례에서 간 생검으로 확인)이 있었습니다.
치료 첫 3개월 동안은 2주마다, 그 후에는 매달, 그리고 임상적으로 필요시에 더 자주 ALT, AST, 총 빌리루빈 등 간기능 검사를 실시하여 모니터링하십시오. 약물 이상반응의 중증도에 따라 표 3에 설명된 대로 ALECENSA 투여를 중단하고 감량 투여를 재개하거나 영구 중단을 하십시오 [투여량 및 투여방법 (2.4)을 참조하십시오].
5.2 간질성 폐질환(ILD)/폐렴
ALECENSA로 치료받은 환자에서 ILD/폐렴이 발생했습니다.
통합 안전성 집단에서[이상반응 (6.1)을 참조하십시오] ALECENSA로 치료받은 환자의 1.3%에서 ILD/폐렴이 발생했고, 0.4%의 환자에서 3등급 ILD/폐렴이 발생했습니다.
통합 안전성 집단에서 5명의 환자(0.9%)가 ILD/폐렴으로 인해 ALECENSA 투여를 중단했습니다. 3등급 이상 ILD/폐렴의 발병 중앙값은 2.1개월(범위: 0.6개월~3.6개월)이었습니다.
ILD/폐렴 징후(예: 호흡곤란, 기침, 발열)가 나타나는 모든 환자에 대해 ILD/폐렴의 가능성을 신속하게 조사하십시오. ILD/폐렴으로 진단된 환자에서는 즉시 ALECENSA 투여를 중단하고, 다른 ILD/폐렴 원인이 발견되지 않으면 영구 중단하십시오[투여량 및 투여방법 (2.4) 및 이상반응 (6)을 참조하십시오].
5.3 신장애
ALECENSA로 치료받은 환자에서 치명적인 사례를 포함한 신장애가 발생했습니다.
통합 안전성 집단에서[이상반응 (6.1)을 참조하십시오] ALECENSA로 치료받은 환자의 12%에서 신장애가 발생했고, 1.7%의 환자에서 3등급 이상, 그 중 0.4%는 치명적이었습니다. 3등급 이상 신장애의 발생 중앙 시간은 3.7개월(범위 0.5~31.8개월)이었습니다. 환자의 2.4%에서 신장애로 인한 투여량 조절이 필요했습니다.
4등급 신장 독성이 있으면 ALECENSA를 영구 중단하십시오. 3등급 신장 독성이 있으면 신기능이 정상 상한치의 1.5배 이하로 회복될 때까지 ALECENSA 투여를 중단한 후 감량하여 재개하십시오[투여량 및 투여방법 (2.4)를 참조하십시오].
5.4 서맥
ALECENSA로 치료받은 환자에서 증상성 서맥이 발생했습니다.
통합 안전성 집단에서[이상반응 (6.1)을 참조하십시오] ALECENSA로 치료받은 환자의 11%에서 서맥이 발생했습니다. 연속 심전도 데이터가 있었던 521명의 ALECENSA 투여 환자 중 20%에서 투여 후 심박수가 분당 50회 미만으로 떨어졌습니다.
심박수와 혈압을 정기적으로 모니터링하십시오. 무증상 서맥인 경우에는 투여량 조절이 필요하지 않습니다. 생명을 위협하지 않는 증상성 서맥인 경우에는 회복될 때까지 ALECENSA 투여를 중단하고, 서맥을 유발할 수 있는 병용 약물 및 고혈압 치료제를 검토하십시오. 병용 약물로 인한 서맥으로 판단되면 무증상 서맥이나 심박수가 분당 60회 이상으로 회복된 후 ALECENSA를 감량 투여로 재개하고 임상적으로 필요한 대로 자주 모니터링하십시오.
생명을 위협하는 서맥 발생 시 기여 요인이 되는 병용 약물이 없다면 ALECENSA를 영구 중단하십시오[투여량 및 투여방법 (2.4)를 참조하십시오]. 생명을 위협하는 서맥이 재발하면 ALECENSA를 영구 중단하십시오.
5.5 중증 근육통 및 크레아틴 인산화 효소(CPK) 상승
ALECENSA로 치료받은 환자에서 중증 근육통과 크레아틴 인산화 효소(CPK) 상승이 발생했습니다.
통합 안전성 집단에서[이상반응 (6.1)을 참조하십시오] ALECENSA로 치료받은 환자의 31%에서 근육통(근육 및 근골격계 관련 반응 포함)이 발생했고, 0.8%의 환자에서 3등급 이상이었습니다. 근육통 사례로 인한 투여량 조절이 환자의 2.1%에서 필요했습니다.
CPK 실험실 데이터가 있었던 491명의 환자 중 통합 안전성 집단에서 ALECENSA로 치료받은 환자의 56%에서 CPK 상승이 있었고, 6%에서 3등급 이상이었습니다. 3등급 이상 CPK 상승의 중앙 발생 시점은 15일(사분위수 범위 – 15~337일)이었습니다. CPK 상승으로 인한 투여량 조절이 환자의 5%에서 필요했습니다.
ALINA 연구에서 CPK 실험실 데이터가 있었던 128명 중 77%의 환자에서 CPK 상승이 있었고, 6%에서 3등급 이상의 상승이 있었습니다.
설명되지 않는 근육통, 압통, 근력 약화 증상이 있으면 환자에게 보고하도록 하십시오. 치료 첫 1개월 동안은 2주마다, 그리고 증상을 보고한 환자에서는 임상적으로 필요한 대로 CPK 수치를 확인하십시오. CPK 상승의 중증도에 따라 ALECENSA 투여를 중단한 후 재개하거나 감량하십시오[투여량 및 투여방법 (2.4)를 참조하십시오].
5.6 용혈성 빈혈
ALECENSA로 치료받은 환자에서 용혈성 빈혈이 발생했습니다.
용혈성 빈혈은 ALECENSA를 투여 받은 환자에서 시판 후 초기에 보고되었으며, 직접 항글로불린 검사(DAT) 결과가 음성인 경우도 있었습니다. 용혈성 빈혈 여부를 확인하기 위한 검사가 ALINA 연구에서 수행되었고, 용혈성 빈혈이 ALECENSA 투여 환자의 3.1%에서 관찰되었습니다. 용혈성 빈혈이 의심되면 ALECENSA 투여를 중단하고 적절한 실험실 검사를 실시하십시오. 용혈성 빈혈이 확인되면, 증상이 해소된 후 감량된 용량으로 투여 재개를 고려하거나 ALECENSA를 영구 중단하십시오 [투여 및 투여량(2.4) 참조].
5.7 태아 독성
동물 연구 결과와 작용 기전을 바탕으로, ALECENSA는 임신부에게 투여 시 태아에게 해를 줄 수 있습니다. 기관 형성기에 임신 랫트와 토끼에 alectinib을 경구 투여한 결과, 모체 독성 용량에서 사람에서 관찰된 노출량의 약 2.7배 수준에서 태아 독성과 유산이 나타났습니다. 임신부와 가임 여성에게 태아에 대한 잠재적 위험성을 알리십시오.
가임 여성에게 ALECENSA 투여 중과 마지막 투여 후 5주 동안 효과적인 피임법을 사용하도록 권고하십시오 [특정 집단에서의 사용(8.1 및 8.3), 임상 약리학(12.1) 참조].
6. 부작용
다음의 이상반응은 라벨의 다른 부분에서 더 자세히 설명되어 있습니다:
- 간독성 [경고 및 주의사항(5.1)참조]
- 간질성 폐질환(ILD)/폐렴 [경고 및 주의사항(5.2)참조]
- 신장 손상 [경고 및 주의사항(5.3)참조]
- 서맥 [경고 및 주의사항(5.4)참조]
- 심한 근육통과 크레아틴 인산 활성 효소(CPK) 상승 [경고 및 주의사항(5.5)참조]
- 용혈성 빈혈 [경고 및 주의사항(5.6)참조]
- 태아에 대한 독성 [경고 및 주의사항(5.7)참조]
6.1 임상시험 경험
임상시험은 광범위한 조건 하에서 수행되므로, 한 약물의 임상시험에서 관찰된 이상반응 발생률을 다른 약물의 임상시험에서 관찰된 발생률과 직접 비교할 수 없으며, 또한 실제 의료 환경에서 관찰되는 발생률을 반영하지 못할 수 있습니다.
경고 및 주의사항에 설명된 통합 안전성 집단은 연구 NP28761, NP28673, ALEX 및 ALINA에서 단일 제제로서 1일 2회 600mg의 경구 알렉센사 투여로 533명의 환자에게 노출된 것을 반영합니다 [임상시험 (14)참조]. 알렉센사 투여환자 533명 중 75%는 6개월 이상, 64%는 1년 이상 투여되었습니다. 이 통합 안전성 집단에서, 가장 흔한 (≥ 20%) 이상반응은 간독성(41%), 변비(39%), 피로(36%), 근육통(31%), 부종(29%), 발진(23%) 및 기침(21%)이었습니다. 가장 흔한 (≥ 2%) Grade 3 또는 4 실험실 이상은 CPK 증가(6%), 혈색소 감소(4.4%), ALT 증가(4.2%), 빌리루빈 증가(4.0%) 및 AST 증가(3.4%)였습니다.
절제된 ALK-양성 비소세포폐암의 보조 치료
알렉센사의 안전성은 절제된 ALK-양성 비소세포폐암 환자의 보조 치료를 위한 다기관, 공개 라벨, 무작위 배정 시험 ALINA에서 평가되었습니다 [임상시험 (14.1)참조]. 무병 생존 분석 시점에서, 알렉센사 투여군의 중앙 노출 기간은 23.9개월이었고 백금 기반 화학요법군은 2.1개월이었습니다.
알렉센사 투여군의 13%에서 중대한 이상반응이 발생했으며, 가장 흔한 중대한 이상반응(≥ 1%)은 폐렴(3.9%), 충수염(3.1%) 및 급성 심근경색(1.6%)이었습니다. 이상반응으로 인한 알렉센사의 영구 중단은 5%의 환자에서 발생했으며, 가장 흔한 중단 이유(≥ 1%)는 폐렴 및 간독성이었습니다.
이상반응으로 인한 알렉센사 투여 중단은 27%의 환자에서 발생했습니다. 투여 중단이 필요한 이상반응(≥ 2% 환자)에는 간독성, CPK 증가, COVID-19, 근육통, 복통 및 폐렴이 포함되었습니다.
이상반응으로 인한 알렉센사 용량 감소는 26%의 환자에서 발생했습니다. 용량 감소가 필요한 이상반응(≥ 2% 환자)에는 간독성, CPK 증가, 발진, 서맥 및 근육통이 포함되었습니다.
표 4 및 5는 ALINA에서 관찰된 흔한 이상반응과 실험실 이상을 요약합니다.
| 이상반응 | 알렉센사 N=128 |
화학요법 N=120 |
||
|---|---|---|---|---|
| 전체 등급(%) | 3-4등급(%) | 전체 등급(%) | 3-4등급(%) | |
| NCI CTCAE v5.0 기준 | ||||
|
||||
| 간담도계 장애 | ||||
| 간독성* | 61 | 4.7† | 13 | 0 |
| 위장관계 장애 | ||||
| 변비 | 42 | 0.8† | 25 | 0.8† |
| 복통‡ | 13 | 0 | 10 | 1.7† |
| 설사§ | 13 | 0.8† | 9 | 1.7† |
| 근골격계 | ||||
| 근육통¶ | 34 | 0.8† | 1.7 | 0 |
| 감염 및 기생충 침입 | ||||
| COVID-19 | 29 | 0 | 0.8 | 0 |
| 전신 및 투여 부위 장애 | ||||
| 피로# | 25 | 0.8† | 28 | 4.2† |
| 부종Þ | 16 | 0 | 1.7 | 0 |
| 피부 및 피하조직 장애 | ||||
| 발진ß | 23 | 1.6† | 10 | 0 |
| 호흡기계 장애 | ||||
| 기침à | 20 | 0.8† | 3.3 | 0 |
| 호흡곤란è | 13 | 0.8† | 2.5 | 0 |
| 신장 장애 | ||||
| 신장 장애ð | 16 | 0.8† | 9 | 0 |
| 신경계 장애 | ||||
| 미각장애ø | 13 | 0 | 3.3 | 0 |
| 두통 | 11 | 0 | 7 | 0 |
| 검사 결과 | ||||
| 체중 증가 | 13 | 0.8† | 0.8 | 0 |
| 심장 장애 | ||||
| 서맥ý | 12 | 0 | 0 | 0 |
| 변수 | ALECENSA N=128 |
화학요법 N=120 |
||
|---|---|---|---|---|
| 전체 등급 (%) | 3-4등급 (%) | 전체 등급 (%) | 3-4등급 (%) | |
| NCI CTCAE v5.0 기준 | ||||
|
||||
| 화학 | ||||
| CPK 증가 | 77 | 8 | 8 | 1.7* |
| AST 증가 | 75 | 0.8* | 25 | 0 |
| 빌리루빈 증가 | 68 | 2.3* | 4.2 | 0 |
| 알칼리성 인산가수분해효소 증가 | 64 | 0 | 14 | 0 |
| ALT 증가 | 57 | 2.3* | 28 | 0 |
| 크레아티닌 증가 | 41 | 0 | 23 | 0 |
| 요산 증가 | 30 | 0 | 19 | 0 |
| 혈액학 | ||||
| 헤모글로빈 감소 | 69 | 0 | 67 | 0.8* |
이전에 치료받지 않은 전이성 ALK-양성 비소세포폐암
ALECENSA의 안전성은 ALEX 연구에서 ALK-양성 비소세포폐암 환자 152명을 대상으로 평가되었습니다. ALECENSA 노출 기간의 중앙값은 17.9개월이었습니다. ALEX 연구 집단(n=303)의 환자 특성은 다음과 같습니다: 중앙 연령 56세, 65세 미만(77%), 여성(56%), 백인(50%), 아시아인(46%), 선암종 조직학(92%), 비흡연(63%), ECOG PS 0 또는 1(93%).
ALECENSA 치료 환자 28%에서 중대한 이상반응이 발생했으며, ALECENSA 치료 환자 2% 이상에서 보고된 중대한 이상반응은 폐렴(4.6%)과 신장애(3.9%)였습니다. ALECENSA 군 환자의 41%에서 3등급 이상의 이상반응이 보고되었습니다. ALECENSA 치료 환자 3.3%에서 치명적인 이상반응이 발생했으며, 이는 신장애(2명), 급사, 심정지 및 폐렴(각각 1명)이었습니다. 이상반응으로 인한 ALECENSA 영구 중단이 환자의 11%에서 발생했습니다. 1% 이상의 환자에서 ALECENSA 중단으로 이어진 약물이상반응은 신장애(2.0%), 고빌리루빈혈증(1.3%), ALT 증가(1.3%), AST 증가(1.3%)였습니다. 이상반응으로 인한 ALECENSA의 투여 중단은 환자의 20%에서 발생했습니다. 2% 이상의 환자에서 투여 중단이 필요했던 이상반응에는 ALT 증가, 폐렴이 포함되었습니다. 이상반응으로 인한 ALECENSA의 용량 감소는 환자의 17%에서 발생했습니다. 2% 이상의 환자에서 용량 감소가 필요했던 이상반응에는 고빌리루빈혈증, AST 증가 및 ALT 증가가 포함되었습니다.
표 6과 표 7은 ALEX에서 관찰된 일반적 이상반응과 실험실 검사치 이상을 요약합니다.
| 이상반응 | ALECENSA N=152 |
Crizotinib N=151 |
||
|---|---|---|---|---|
| 전체 등급 (%) | 3-4등급 (%) | 전체 등급 (%) | 3-4등급 (%) | |
| NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; MedDRA = Medical Dictionary for Regulatory Activities; SOC = System Organ Class. | ||||
| 위장관 | ||||
| 변비 | 34 | 0 | 33 | 0 |
| 구역질 | 14 | 0.7 | 48 | 3.3 |
| 설사 | 12 | 0 | 45 | 2.0 |
| 전신 | ||||
| 피로* | 26 | 1.3 | 23 | 0.7 |
| 부종† | 22 | 0.7 | 34 | 0.7 |
| 근골격계 | ||||
| 근육통‡ | 23 | 0 | 4.0 | 0 |
| 피부 | ||||
| 발진§ | 15 | 0.7 | 13 | 0 |
| 심장 | ||||
| 서맥¶ | 11 | 0 | 15 | 0 |
| 신장 | ||||
| 신장 장애# | 12 | 3.9Þ | 0 | 0 |
| 매개변수 | ALECENSA N=152 |
Crizotinib N=151 |
||
|---|---|---|---|---|
| 전체 등급 (%) | 3-4 등급 (%) | 전체 등급 (%) | 3-4 등급 (%) | |
| 참고: 국립암연구소 공통독성평가기준 v4.03 기반. 기저시점 실험실 평가가 없는 환자는 제외됨. | ||||
|
||||
| 화학검사 | ||||
| 과빌리루빈혈증* | 54 | 5 | 4.7 | 0 |
| AST 증가† | 50 | 6 | 56 | 11 |
| 알칼리인산가수분해효소 증가‡ | 50 | 0 | 44 | 0 |
| ALT 증가‡ | 40 | 6 | 62 | 16 |
| 크레아티닌 증가‡,§ | 38 | 4.1 | 23 | 0.7 |
| CPK 증가¶ | 37 | 2.8 | 52 | 1.4 |
| 저칼슘혈증* | 29 | 0 | 61 | 1.4 |
| 고혈당증# | 22 | 2.2 | 19 | 2.3 |
| 저나트륨혈증Þ | 18 | 6 | 20 | 4.1 |
| 저칼륨혈증‡ | 17 | 2 | 12 | 0.7 |
| 저알부민혈증ß | 14 | 0 | 57 | 3.4 |
이전에 크리조티닙으로 치료받은 전이성 ALK-양성 비소세포폐암
ALECENSA의 안전성은 두 가지 임상시험인 NP28761 및 NP28673에서 ALECENSA로 치료받은 253명의 ALK-양성 비소세포폐암 환자들을 대상으로 평가되었습니다. ALECENSA 노출의 중간 기간은 9.3개월이었습니다. 169명의 환자(67%)가 6개월 이상 ALECENSA에 노출되었고, 100명의 환자(40%)가 1년 이상 노출되었습니다. 인구 특성은 다음과 같습니다: 중간 연령 53세, 65세 미만(86%), 여성(55%), 백인(74%), 아시아인(18%), 비소세포폐암 선암종 조직 유형(96%), 비흡연 또는 과거 흡연 경험(98%), ECOG 수행능력 상태 0 또는 1(91%), 이전 항암화학요법 치료(78%)였습니다.
19%의 환자에서 중대한 이상반응이 발생했으며, 가장 빈번하게 보고된 중대한 이상반응은 폐색전증(1.2%), 호흡곤란(1.2%), 고빌리루빈혈증(1.2%)이었습니다. 2.8%의 환자에서 치명적인 이상반응이 발생했으며 이는 출혈(0.8%), 장천공(0.4%), 호흡곤란(0.4%), 폐색전증(0.4%), 심내막염(0.4%)을 포함했습니다. 6%의 환자에서 ALECENSA 치료를 이상반응으로 인해 영구 중단했습니다. 영구 중단의 가장 흔한 원인 이상반응은 고빌리루빈혈증(1.6%), ALT 증가(1.6%), AST 증가(1.2%)였습니다. 전반적으로, 권장 용량으로 치료를 시작한 환자의 23%에서 최소한 1회의 용량 감소가 필요했습니다. 첫 번째 용량 감소까지의 중간 시간은 48일이었습니다. 용량 감소나 중단의 가장 흔한 원인은 빌리루빈 증가(6%), CPK 증가(4.3%), ALT 증가(4.0%), AST 증가(2.8%), 구토(2.8%)였습니다.
표 8과 표 9는 NP28761 및 NP28673 연구에서 관찰된 일반적인 이상반응과 실험실 수치 이상을 요약하고 있습니다.
| 이상반응 | ALECENSA N=253 |
|
|---|---|---|
| 모든 등급(%) | 3-4등급(%)* | |
| 피로감† | 41 | 1.2 |
| 변비 | 34 | 0 |
| 부종‡ | 30 | 0.8 |
| 근육통§ | 29 | 1.2 |
| 기침 | 19 | 0 |
| 발진¶ | 18 | 0.4 |
| 메스꺼움 | 18 | 0 |
| 두통 | 17 | 0.8 |
| 설사 | 16 | 1.2 |
| 호흡곤란 | 16 | 3.6# |
| 요통 | 12 | 0 |
| 구토 | 12 | 0.4 |
| 체중 증가 | 11 | 0.4 |
| 시력 장애Þ | 10 | 0 |
추가적인 임상적으로 중요한 약물 이상반응은 광과민증으로, 연구 NP28761 및 NP28673에서 ALECENSA에 노출된 환자의 9.9%에서 발생했습니다. 환자들은 햇빛 노출을 피하고 광범위 스펙트럼 자외선 차단제를 사용하도록 조언받았습니다. 2등급 광과민증의 발생률은 0.4%였고, 나머지 사례는 1등급의 중증도였습니다.
8 특정 인구집단에서의 사용
8.1 임신
위험 요약
동물 연구 결과와 작용기전을 근거로 ALECENSA는 임신부에게 투여할 경우 태아에게 해를 끼칠 수 있습니다 [임상 약리학(12.1) 참조]. 임신부에서 ALECENSA 사용에 대한 자료는 없습니다.
임신 기간 중 랫드와 토끼에게 경구 투여한 알렉티닙은 모체 독성 용량에서 사람에게 알렉티닙 600mg을 하루 두 번 투여했을 때 관찰되는 노출의 약 2.7배에 해당하는 노출에서 태아 독성과 유산을 초래했습니다 (아래 Data 참조). 임신부에게 태아에 대한 잠재적 위험을 알리십시오.
미국 일반 인구에서 임상적으로 확인된 임신 시 주요 선천성 기형 및 유산의 예상 배경 위험률은 각각 2~4% 및 15~20%입니다.
자료
동물 자료
예비 토끼 태아-배아 연구에서 기관 형성기에 경구 투여한 알렉티닙은 모체 독성 용량 27mg/kg/day(사람에서 알렉티닙 600mg을 하루 두 번 투여 시 예상 전신 노출의 약 2.9배)에서 임신 토끼 6마리 중 3마리에서 유산 또는 완전 태아-배아 치사를 초래했습니다. 이 그룹의 나머지 3마리 임신 토끼는 생존 태아가 적었고 태아와 태반 중량이 감소했으며, 후식도 쇄골하동맥이 관찰되었습니다. 예비 랫드 배아-태아 발달 연구에서 기관 형성기에 알렉티닙 투여 시 27mg/kg/day(사람에서 알렉티닙 600mg을 하루 두 번 투여 시 예상 전신 노출의 약 4.5배)에서 모든 임신 랫드에서 배자 전체 소실이 발생했습니다. 9mg/kg/day 이상의 용량(사람에서 알렉티닙 600mg을 하루 두 번 투여 시 예상 전신 노출의 약 2.7배)에서 모체 독성과 태아 중량 감소, 요관 확장, 흉선경, 소심실 및 심실벽 얇아짐, 천골 및 미골 척추 수 감소 등의 발달 독성이 관찰되었습니다.
8.3 생식능력이 있는 여성 및 남성
ALECENSA는 임신부에게 투여할 경우 태아에게 해를 끼칠 수 있습니다 [특정 인구집단에서의 사용(8.1) 참조].
피임
남성
유전 독성 결과를 기반으로 생식능력이 있는 여성 파트너가 있는 남성에게 ALECENSA 치료 중 및 마지막 투여 후 3개월 동안 효과적인 피임법을 사용하도록 조언하십시오 [비임상 독성(13.1) 참조].
8.5 노인 사용
NP28761, NP28673, ALEX 및 ALINA 연구에 참여한 533명 환자 중 19%가 65세 이상(3.2%는 75세 이상)이었습니다. 연령에 따른 전반적인 유효성 차이는 관찰되지 않았습니다. 탐색적 분석 결과 65세 이상 환자에서 중대한 이상반응(38% vs 25%), 치료 중단을 초래한 이상반응(18% vs 6%), 용량 조절(48% vs 35%)이 65세 미만 환자에 비해 더 자주 발생했습니다.
8.6 신장애
경증 또는 중등증 신장애 환자에서 용량 조절은 권장되지 않습니다. 중증 신장애(크레아티닌 청소율 30mL/min 미만) 또는 말기 신질환 환자에서 ALECENSA의 안전성은 연구된 바 없습니다 [임상 약리학(12.3) 참조].
8.7 간장애
경증(Child-Pugh A) 또는 중등증(Child-Pugh B) 간장애 환자에서 용량 조절은 권장되지 않습니다. 중증 간장애(Child-Pugh C) 환자에서 알렉티닙 노출이 증가했습니다. 중증 간장애(Child-Pugh C) 환자에서 ALECENSA 권장 용량은 하루 450mg씩 두 번 경구 투여입니다 [용법 및 투여량(2.3), 임상 약리학(12.3) 참조].
10 과다 복용
과량 투여에 대한 경험은 없습니다. ALECENSA 과량 투여에 대한 특정 해독제는 없습니다. Alectinib 및 주요 활성 대사체 M4는 혈장 단백질에 99% 이상 결합되어 있으므로, 혈액 투석은 과량 투여 치료에 효과적이지 않을 것으로 예상됩니다.
11 설명
ALECENSA(알렉티닙)는 경구 투여용 키나제 억제제입니다. 알렉티닙의 분자식은 C30H34N4O2 ∙ HCl입니다. 분자량은 482.62 g/mol(프리베이스 형태)이고 519.08 g/mol(염산염)입니다. 알렉티닙은 9-에틸-6, 6-디메틸-8-[4-(모르폴린-4-일)피페리딘-1-일]-11-옥소-6, 11-디하이드로-5H-벤조[b]카바졸-3-카르보니트릴 염산염으로 화학적으로 기술됩니다. 알렉티닙의 화학 구조는 아래와 같습니다.
알렉티닙 HCl은 pKa가 7.05(베이스)인 흰색에서 노란 흰색 가루 또는 덩어리 가루입니다.
ALECENSA는 150mg의 알렉티닙(알렉티닙 HCl 161.33mg에 해당)과 다음과 같은 비활성 성분들로 이루어진 경질 캡슐 제제입니다: 무수 유당, 하이드록시프로필셀룰로오스, 나트륨 라우릴황산염, 스테아린산 마그네슘 및 카르복시메틸셀룰로오스 칼슘. 캡슐 셸은 하이프로멜로오스, 카라기난, 염화칼륨, 이산화티탄, 옥수수전분 및 카르노바 왁스로 구성되어 있습니다. 인쇄용 잉크는 적색산화철(E172), 황색산화철(E172), FD&C 청색 No. 2 알루미늄레이크(E132), 카르노바 왁스, 백셀락 및 글리세릴 모노올레에이트로 이루어져 있습니다.
12 임상 약리학
12.1 작용 기전
Alectinib은 ALK와 RET를 타겟으로 하는 타이로신 키나아제 억제제입니다. 비임상 연구에서 alectinib은 ALK 인산화와 하위신호전달 단백질 STAT3 및 AKT의 ALK 매개 활성화를 억제하고, ALK 융합, 증폭 또는 활성화 돌연변이를 가진 여러 세포주에서 종양 세포 생존력을 감소시켰습니다. Alectinib의 주요 활성 대사체인 M4도 유사한 시험관 내 효능과 활성을 보였습니다.
Alectinib과 M4는 시험관 내와 생체 내에서 crizotinib에 내성이 발현된 NSCLC 환자에게서 발견된 여러 ALK 효소 돌연변이 형태를 포함한 다양한 돌연변이형 ALK에 대해 활성을 나타냈습니다.
ALK 융합을 가진 종양을 이식한 마우스 모델에서 alectinib 투여는 항종양 활성과 생존기간 연장을 보였으며, ALK 의존성 종양 세포주를 뇌 내에 이식한 모델에서도 마찬가지였습니다.
12.3 약동학
Alectinib과 주요 활성 대사체 M4의 약동학은 ALK-양성 NSCLC 환자와 건강한 피험자에게서 특성화되었습니다.
ALK-양성 NSCLC 환자에서 alectinib의 기하평균(변동계수 %) 정상상태 최대농도(Cmax,ss)는 665 ng/mL(44%), M4는 246 ng/mL(45%)였고, 최고농도와 최저농도 비율은 1.2였습니다. Alectinib의 기하평균 정상상태 0-12시간 농도곡선하면적(AUC0-12h,ss)은 7,430 ng*h/mL(46%), M4는 2,810 ng*h/mL(46%)였습니다. Alectinib 노출량은 섭취 후 460 mg에서 900 mg 용량 범위(승인된 권장 용량의 0.75~1.5배)에서 용량 비례성을 보였습니다. Alectinib과 M4는 7일차에 정상상태 농도에 도달했고, 기하평균 축적율은 둘 다 약 6배였습니다.
흡수
ALK-양성 NSCLC 환자에서 ALECENSA 600 mg을 1일 2회 투여 시 섭취 후 4시간에 alectinib 최대농도에 도달했습니다.
Alectinib의 절대 생체이용률은 섭취 후 37%(90% CI: 34%, 40%)였습니다.
고지방, 고칼로리 식사는 ALECENSA 600 mg 단회 경구투여 후 alectinib과 M4의 전체 노출량(AUC0-inf)을 3.1배(90% CI: 2.7, 3.6) 증가시켰습니다.
분포
Alectinib과 M4의 겉보기 분포용적은 각각 4,016 L, 10,093 L입니다.
Alectinib과 M4는 농도에 관계없이 인체 혈장 단백질에 99% 이상 결합합니다.
ALK-양성 NSCLC 환자의 뇌척수액 내 alectinib 농도는 혈장 내 추정 유리 alectinib 농도에 근접합니다.
In vitro 연구에 따르면 alectinib은 P-glycoprotein(P-gp)의 기질은 아니지만, M4는 P-gp의 기질입니다. Alectinib과 M4는 breast cancer resistance protein(BCRP), organic anion-transporting polypeptide(OATP) 1B1 또는 OATP1B3의 기질은 아닙니다.
제거
겉보기 클리어런스(CL/F)는 alectinib의 경우 81.9 L/시간, M4는 217 L/시간입니다. ALK-양성 NSCLC 환자에서 alectinib과 M4의 기하평균 반감기는 각각 33시간, 31시간입니다.
특정 집단
연령(21~83세), 체중(38~128kg), 경증 간장애(총 빌리루빈 ≤ ULN 및 AST > ULN 또는 총 빌리루빈 1~≤ 1.5 × ULN 및 AST 모든 값), 경증~중등도 신장애(크레아티닌 클리어런스 30~89 mL/min), 인종(백인, 아시아인, 기타), 성별은 alectinib과 M4의 전신 노출량에 임상적으로 의미 있는 영향을 미치지 않았습니다. 중증 신장애(크레아티닌 클리어런스 < 30 mL/min) 또는 말기 신부전 환자에서의 alectinib 약동학은 연구되지 않았습니다.
간장애: ALECENSA 300 mg 단회 경구 투여 후, 정상 간기능 피험자 대비 중등도 간장애(Child-Pugh B) 피험자의 alectinib과 M4의 전체 AUCinf 기하평균비[90% 신뢰구간]는 1.36[0.947, 1.96], 중증 간장애(Child-Pugh C) 피험자는 1.76[0.984, 3.15]이었습니다. 세 군간 alectinib과 M4의 전체 Cmax는 유사했습니다. 경증 또는 중등도 간장애 환자에서 용량 조절은 필요하지 않습니다. 중증 간장애 환자에 대한 ALECENSA의 권장 용량은 1일 2회, 경구 450mg입니다 [용법 용량(2.3) 및 특정 집단에서의 사용(8.7)항 참조].
약물 상호작용
다른 약물이 alectinib에 미치는 영향
강력한 CYP3A 억제제(posaconazole), 강력한 CYP3A 유도제(rifampin) 또는 위산 분비 억제제(esomeprazole)와 병용 시, 임상 연구에서 alectinib과 M4의 전체 노출량에 임상적으로 의미 있는 변화는 관찰되지 않았습니다.
Alectinib이 다른 약물에 미치는 영향
ALECENSA와 병용 시 midazolam(민감한 CYP3A 기질) 또는 repaglinide(민감한 CYP2C8 기질)의 노출량이 임상적으로 의미있게 변화할 것으로 예상되지 않습니다.
시험관 내 연구에 따르면 alectinib과 M4는 CYP1A2, 2B6, 2C9, 2C19 또는 2D6를 억제하지 않습니다.
시험관 내 연구에 따르면 alectinib과 M4는 P-gp와 BCRP를 억제합니다. Alectinib은 OATP1B1, OATP1B3, OAT1, OAT3 또는 OCT2 수송활성을 억제하지 않습니다.
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식능력 손상
알렉티닙의 발암성 연구는 수행되지 않았습니다.
알렉티닙은 in vitro 박테리아 복귀 돌연변이 (에임스) 분석에서는 돌연변이 유발성이 없었지만 랫트 골수 소핵 시험에서 소핵 수의 증가로 양성 반응을 보였습니다. 소핵 유도 기전은 염색체 이상 분리 (비분리성)이었으며 염색체에 대한 분열 유발성 효과는 아니었습니다.
동물 연구에서 알렉티닙이 생식능력에 미치는 영향을 평가한 적은 없습니다. 랫트와 원숭이에서 수행된 일반 독성 연구에서 수컷과 암컷 생식기관에 대한 부작용은 관찰되지 않았습니다.
14 임상 연구
14.1 절제된 ALK-양성 NSCLC의 보조요법
완전한 종양 절제술 후 ALK-양성 NSCLC 환자의 보조요법 치료로서 ALECENSA의 유효성은 글로벌 무작위 공개 임상시험(ALINA: NCT03456076)에서 평가되었습니다. 적격 환자는 절제 가능한 ALK-양성 NSCLC로 Union for International Cancer Control/American Joint Committee on Cancer(UICC/AJCC) Staging System, 7판에 따라 병기 IB(종양 ≥ 4cm) – IIIA여야 했습니다. ALK 재배열은 현지에서 실시하는 FDA 승인 ALK 검사 또는 중앙에서 실시하는 VENTANA ALK(D5F3) CDx 검사로 확인했습니다.
무작위 배정은 인종(아시아인 vs. 기타 인종) 및 병기(IB vs. II vs. IIIA)로 층화되었습니다. 환자는 종양 절제술 후 ALECENSA 600mg을 하루 2차례 경구 투여하거나 화학요법제로 무작위 배정(1:1)받았습니다. ALECENSA 치료는 2년간 지속되거나 질병 재발 또는 허용할 수 없는 독성이 있을 때까지 계속했습니다. 화학요법제는 아래와 같은 1개 레지멘으로 정맥 투여되었으며 주기당 21일, 총 4주기 투여했습니다.
- Cisplatin 75 mg/m2(1일차) 및 vinorelbine 25 mg/m2(1 및 8일차)
- Cisplatin 75 mg/m2(1일차) 및 gemcitabine 1250 mg/m2(1 및 8일차)
- Cisplatin 75 mg/m2(1일차) 및 pemetrexed 500 mg/m2(1일차)
Cisplatin 기반 요법을 견디기 힘들 경우 carboplatin으로 대체하여 위 병용요법에 AUC 5 mg/mL/min 또는 6 mg/mL/min 용량으로 투여했습니다.
주요 유효성 평가변수는 병기 II-IIIA NSCLC 환자 및 병기 IB-IIIA NSCLC 환자(실제 치료 집단[ITT])에서 연구자 평가 무병 생존기간(DFS)이었습니다. DFS는 무작위 배정일부터 첫 번째 문서화된 질병 재발, 새로운 일차 NSCLC 발생, 또는 어떤 원인으로든 사망 중 첫 번째 사유의 발생일까지의 기간으로 정의되었습니다. 추가 유효성 평가변수는 ITT 집단의 전체 생존기간(OS)이었습니다.
총 257명의 환자가 ALECENSA군(130명) 또는 화학요법군(127명)에 무작위 배정되었습니다. 중앙연령은 56세(범위: 26~87세), ≥ 65세가 24%였으며 52%는 여성, 56%는 아시아인, 42%는 백인, 0.4%는 흑인 또는 아프리카계 미국인, 2.3%는 인종 불명이었으며 0.4%는 히스패닉 또는 라티노였습니다. 60%가 비흡연자였으며 53%가 ECOG 활동수행 점수가 0이었습니다. 병기 IB가 10%, II가 35%, IIIA가 55%였습니다.
ALINA 시험에서 ALECENSA 치료군이 화학요법군에 비해 DFS 유의한 개선을 보였습니다. 이 때 OS 데이터는 미성숙 상태였으며 ITT 집단에서 2.3%의 사망만 보고되었습니다.
ALINA의 유효성 결과는 표 10 및 그림 1에 요약되어 있습니다.
| 유효성 평가변수 | 병기 II-IIIA 집단 | ITT 집단 | ||
|---|---|---|---|---|
| ALECENSA N=116 |
화학요법 N=115 |
ALECENSA N=130 |
화학요법 N=127 |
|
| DFS = 무병 생존기간; ITT = 실제 치료 집단; CI = 신뢰구간; NR = 도달하지 않음; NE = 추정할 수 없음. | ||||
| DFS 사례 수(%) | 14 (12) | 45 (39) | 15 (12) | 50 (39) |
| 질병 재발(%) | 14 (12) | 44 (38) | 15 (12) | 49 (38) |
| 사망 | 0 | 1 (0.9) | 0 | 1 (0.8) |
| DFS 중앙값, 개월 (95% CI)* |
NR (NE, NE) |
44.4 (27.8, NE) |
NR (NE, NE) |
41.3 (28.5, NE) |
| 위험비(95% CI)† | 0.24 (0.13, 0.45) | 0.24 (0.13, 0.43) | ||
| p-값‡ | <0.0001 | <0.0001 | ||
그림 1: 알리나(ALINA) 시험에서 연구자에 의해 평가된 무병 생존기간(DFS)의 카플란-마이어 곡선
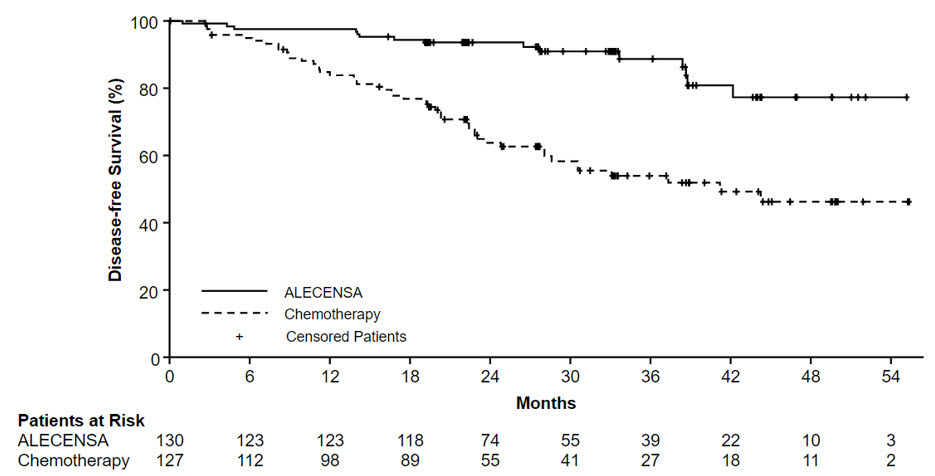
탐색적 분석에서, 질병 재발 시점에서 뇌 전이가 있었던 환자의 비율은 알레센사 군에서 4명(3.1%), 화학요법 군에서 14명(11%)이었다.
14.2 전이성 ALK 양성 비소세포폐암 치료
이전에 치료받지 않은 전이성 ALK 양성 비소세포폐암
이전에 전이성 질병에 대한 전신 요법을 받은 적이 없는 ALK 양성 비소세포폐암 환자의 알레센사 치료 효과는 공개 표지 무작위배정 활성 대조 다기관 연구(ALEX: NCT02075840)를 통해 확립되었다. 환자는 ECOG 수행 상태가 0-2이어야 하고 VENTANA ALK(D5F3) CDx 분석법으로 확인된 ALK 양성 비소세포폐암 상태여야 했다. 치료 여부와 무관하게 안정적 신경학적 상태의 중추신경계(CNS) 전이 환자(뇌실질내 전이 포함)도 가능했으며, CNS 전이 증상이 있는 환자의 경우에는 전신 방사선치료 또는 감마나이프 방사선치료를 등록 최소 14일 전에 받고 임상적으로 안정된 상태여야 했다. 기저 QTc 간격이 470 ms를 초과하는 환자는 제외되었다.
환자는 1:1의 비율로 무작위로 알레센사 600 mg 1일 2회 경구 투여군 또는 크리조티닙 250 mg 1일 2회 경구 투여군에 배정되었다. 무작위 배정에는 ECOG 수행상태(0/1 vs 2), 인종(아시아 vs 기타 인종) 및 기저 시점의 CNS 전이 유무가 층화인자로 사용되었다. 양 군 모두 질병 진행 또는 허용할 수 없는 독성 발생 시까지 치료를 계속하였다. 주요 유효성 평가항목은 RECIST v1.1에 따라 연구자에 의해 평가된 무진행 생존기간(PFS)이었다. 추가 유효성 평가항목으로는 독립평가위원회(IRC)에 의해 평가된 PFS, IRC에 의한 RECIST v1.1 기준 CNS 무진행 기간, 전반적 반응률(ORR), 반응 지속기간(DOR), 전체 생존기간(OS) 등이 있었다. 추가 탐색 평가항목으로는 기저 CNS 전이 환자에서 IRC에 의해 평가된 CNS ORR 및 CNS DOR 등이 있었다.
총 303명의 환자가 알레센사군(n=152) 또는 크리조티닙군(n=151)으로 무작위 배정되었다. 연구 집단의 인구학적 특성은 여성 56%, 중앙연령 56세(범위: 18 – 91세), 백인 50%, 아시안 46%, 흑인 1%, 기타 인종 3%였다. 대부분의 환자가 선암종(92%)이었고 비흡연자(63%)였다. CNS 전이가 있는 환자는 전체의 40%(n=122)였으며, 그 중 43명은 IRC에 의해 측정 가능한 CNS 병변이 있었다. ALEX 연구에서 알레센사군이 유의하게 PFS 개선을 보였다. IRC에 의해 평가한 CNS 진행기간도 알레센사군에서 유의하게 연장되었으며, CNS가 최초 병소 진행인 경우 또는 전신 진행과 동시에 CNS 진행이 발생한 경우가 알레센사군(12%)에 비해 크리조티닙군(45%)에서 높게 관찰되었다. ALEX 연구의 유효성 결과는 표 11 및 그림 2와 같다.
| 알레센사 N=152 |
크리조티닙 N=151 |
|
|---|---|---|
| CNS: 중추신경계, ORR: 전반적 반응률, IRC: 독립평가위원회, CI: 신뢰구간, NE: 추정불가. | ||
| 무진행 생존기간 | ||
| 이벤트 발생 환자 수(%) | 63 (41%) | 92 (61%) |
| 질병 진행(%) | 51 (34%) | 82 (54%) |
| 사망(%) | 12 (8%) | 10 (7%) |
| 중앙값, 개월(95% CI) | 25.7 (19.9, NE) | 10.4 (7.7, 14.6) |
| 위험비(95% CI) * | 0.53 (0.38, 0.73) | |
| P-value * | < 0.0001 | |
| 전반적 반응률 | ||
| 전반적 반응률, %(95% CI) † | 79% (72, 85) | 72% (64, 79) |
| P-value * | 0.1652 | |
| 완전 반응률, % | 13% | 6% |
| 부분 반응률, % | 66% | 66% |
| 반응 지속기간 | ||
| 반응 환자 수 | n=120 | n=109 |
| 6개월 이상 반응 지속 | 82% | 57% |
| ≥12 개월 반응 지속 기간 | 64% | 36% |
| ≥18 개월 반응 지속 기간 | 37% | 14% |
그림 2: ALEX 에서 무진행 생존기간(IRC)의 Kaplan-Meier 곡선
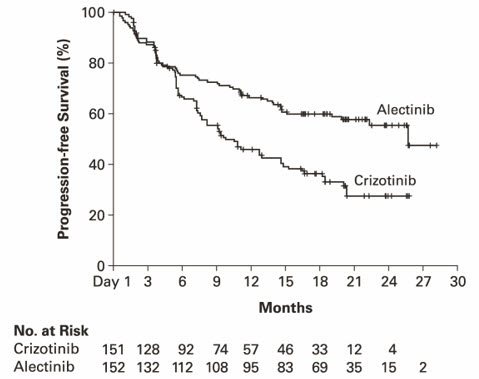
연구자 평가에 의한 PFS 결과(HR=0.48 [95% CI: 0.35-0.66], 층화 로그-순위 p<0.0001)도 IRC와 유사했다. 데이터 커트오프 시점에서 전체 생존 자료는 성숙하지 않았다.
기저 시점에서 측정 가능한 CNS 병변이 있는 환자에서 사전 지정된 탐색적 CNS 반응률 분석 결과는 표 12에 요약되어 있다.
이전에 Crizotinib으로 치료받은 전이성 ALK-양성 NSCLC
ALECENSA의 안전성과 효능은 두 가지 단일군, 다기관 임상시험 NP28761(NCT01588028)과 NP28673(NCT01801111)에서 입증되었습니다. Crizotinib에 진행된 국소 진행성 또는 전이성 ALK-양성 NSCLC 환자, FDA 승인 검사를 통해 ALK-양성 NSCLC가 입증된 환자, ECOG PS 0-2인 환자가 두 연구 모두에 등록되었습니다. 선행 화학요법과 선행 CNS 방사선치료를 받은 환자는 CNS 전이가 최소 2주 동안 안정적이었다면 등록 가능한 선정기준을 충족했습니다. 모든 환자는 경구 ALECENSA 600mg을 1일 2회 투여받았습니다. 두 연구의 주요 효능 평가변수는 독립 평가 위원회(IRC)에 의해 평가된 RECIST v1.1에 따른 객관적 반응률(ORR)이었습니다. IRC에 의해 평가된 추가 평가변수로는 반응 지속 기간(DOR), CNS ORR, CNS DOR이 포함되었습니다.
NP28761은 북미에서 실시되었고 87명의 환자를 등록했습니다. NP28761의 기저 인구통계학적 및 질병 특성으로는 중간 연령 54세(범위 29-79세, 65세 이상 18%), 백인 84%, 아시아인 8%, 여성 55%, ECOG PS 0 35%, ECOG PS 1 55%, 비흡연자 또는 과거 흡연자 100%, Stage IV 99%, 선암 94%, 선행 화학요법 74% 포함되었습니다. 가장 흔한 흉외 전이 부위는 CNS 60%(이 중 65%가 CNS 방사선치료 받음), 림프절 43%, 뼈 36%, 간 34% 등이었습니다.
NP28673은 국제적으로 실시되었고 138명의 환자를 등록했습니다. NP28673의 기저 인구통계학적 및 질병 특성으로는 중간 연령 52세(범위 22-79세, 65세 이상 10%), 백인 67%, 아시아인 26%, 여성 56%, ECOG PS 0 32%, ECOG PS 1 59%, 비흡연자 또는 과거 흡연자 98%, Stage IV 99%, 선암 96%, 선행 화학요법 80% 포함되었습니다. 가장 흔한 흉외 전이 부위는 CNS 61%(이 중 73%가 CNS 방사선치료 받음), 뼈 51%, 림프절 38%, 간 30% 등이었습니다.
NP28761과 NP28673 모든 치료 환자에 대한 효능 결과를 표 13에 요약하였습니다. NP28761 연구의 IRC 및 연구자 평가 모두에서 중간 추적관찰기간은 4.8개월이었고, NP28673 연구의 IRC 평가에서는 10.9개월, 연구자 평가에서는 7.0개월이었습니다. 모든 반응은 부분반응이었습니다.
| 효능 변수 | NP28761 (N=87) | NP28673 (N=138) | ||
|---|---|---|---|---|
| IRC* 평가 | 연구자 평가 | IRC* 평가 | 연구자 평가 | |
|
||||
| 객관적 반응률(95% 신뢰구간) | 38% (28; 49) |
46% (35; 57) |
44% (36; 53) |
48% (39; 57) |
| 반응자 수 | 33 | 40 | 61 | 66 |
| 반응 지속 기간, 중간값 (개월)(95% 신뢰구간) | 7.5 (4.9, 평가불가) |
평가불가 (4.9, 평가불가) |
11.2 (9.6, 평가불가) |
7.8 (7.4, 9.2) |
NP28761과 NP28673 연구에서 RECIST v1.1에 따라 기저치에서 측정 가능한 CNS 병소가 있던 51명의 환자 하위군에 대해 CNS 병소에 대한 ORR과 반응 지속 기간을 평가하였고 그 결과를 표 14에 요약하였습니다. 측정 가능한 CNS 병소를 가진 환자 중 35명(69%)은 과거에 뇌 방사선치료를 받았으며 이 중 25명(49%)은 ALECENSA 투여 시작 최소 6개월 전에 방사선치료를 완료하였습니다. 반응은 과거 방사선치료 유무와 상관없이 관찰되었습니다.
| 효능 변수 | N=51 |
|---|---|
| CNS 객관적 반응률(95% 신뢰구간) | 61% (46, 74) |
| 완전 관해 | 18% |
| 부분 반응 | 43% |
| CNS 반응 지속 기간, 중간값(개월)(95% 신뢰구간) | 9.1 (5.8, 평가불가) |
16 규격/저장 및 취급방법
경질 캡슐, 상단에 “ALE”가 검은색 잉크로 인쇄되고 몸체에 “150 mg”가 검은색 잉크로 인쇄된 흰색 150 mg 캡슐, 구성:
| 병당 240캡슐: | NDC 50242-130-01 |
17 환자 상담 정보
환자에게 FDA 승인 환자 라벨(환자 정보) 읽기를 권고합니다.
환자에게 다음 사항을 알립니다:
간독성
빌리루빈 및 간 아미노전이효소 상승의 징후와 증상을 환자에게 알립니다. 빌리루빈 및 간 아미노전이효소 상승의 징후나 증상이 있을 경우 즉시 의료 전문가에게 연락하도록 환자에게 조언합니다 [경고 및 주의사항(5.1)을 참조하십시오].
간질성 폐렴(ILD)/폐렴
환자에게 중증 ILD/폐렴의 위험을 알립니다. 새로운 호흡기 증상이 나타나거나 악화되면 즉시 의료 전문가에게 연락하도록 환자에게 조언합니다 [경고 및 주의사항(5.2)를 참조하십시오].
신장 장애
중증 및 잠재적으로 치명적일 수 있는 신장 장애의 위험을 환자에게 알립니다. 소변색 변화, 소변량 감소 또는 다리와 발의 부종이 있으면 의료 전문가에게 연락하도록 환자에게 조언합니다 [경고 및 주의사항(5.3)을 참조하십시오].
서맥
어지럼증, 현기증 및 실신과 같은 서맥 증상이 ALECENSA 복용 중에 발생할 수 있음을 환자에게 알립니다. 이러한 증상이 있는 경우 의료 전문가에게 연락하고 심장 또는 혈압 약물 사용에 대해 의료 전문가에게 알리도록 환자에게 조언합니다 [경고 및 주의사항(5.4)를 참조하십시오].
중증 근육통/CPK 상승
근육통, 원인이 설명되지 않거나/그리고 지속적인 근육통, 압통 또는 근력 저하의 징후와 증상을 환자에게 알립니다. 새로운 근육통이나 근력 저하 증상이 나타나거나 악화되면 즉시 의료 전문가에게 연락하도록 환자에게 조언합니다 [경고 및 주의사항(5.5)를 참조하십시오].
광과민증
환자에게 광과민증의 징후와 증상을 알립니다. ALECENSA 복용 중 및 약물 중단 후 최소 7일 동안 장시간 직사광선 노출을 피하고 적절한 자외선 차단을 하도록 환자에게 조언합니다. 환자에게 광범위 자외선 A(UVA)/자외선 B(UVB) 차단제 및 립밤(SPF ≥ 50)을 사용해 잠재적인 화상으로부터 보호하도록 조언합니다 [이상반응(6.1)을 참조하십시오].
배아-태아 독성
ALECENSA는 임신 중 복용하면 태아에게 해를 끼칠 수 있습니다. 임신한 여성과 가임 여성에게 태아에 대한 잠재적 위험을 알립니다 [경고 및 주의사항(5.6) 및 특정 집단에서의 사용(8.1, 8.3)을 참조하십시오].
가임 여성에게 ALECENSA 치료 중과 마지막 ALECENSA 투여 후 5주 동안 효과적인 피임법을 사용하도록 조언합니다. 알려진 임신 또는 의심되는 임신이 있으면 의료 전문가에게 알리도록 환자에게 조언합니다 [경고 및 주의사항(5.6) 및 특정 집단에서의 사용(8.1, 8.3)을 참조하십시오].
가임 여성 파트너가 있는 남성 환자에게 ALECENSA 치료 중 및 마지막 투여 후 3개월 동안 효과적인 피임법을 사용하도록 조언합니다 [특정 집단에서의 사용(8.3) 및 비임상 독성(13.1)을 참조하십시오].
투여
환자에게 ALECENSA를 하루 두 번 복용하도록 지시합니다. 환자에게 음식과 함께 ALECENSA 캡슐을 통째로 삼키도록 조언합니다 [투여량 및 투여방법(2.2)를 참조하십시오].
복용 누락
ALECENSA 복용을 놓치거나 투여 후 구토를 한 경우, 환자에게 추가 복용하지 말고 정상 시간에 다음 복용량을 복용하도록 조언합니다 [투여량 및 투여방법(2.2)를 참조하십시오].
SPL 미분류 섹션
유통업체:
Genentech USA, Inc.
Roche 그룹 회원
1 DNA Way
South San Francisco, CA 94080-4990
ALECENSA®는 Chugai Pharmaceutical Co., Ltd., Tokyo, Japan의 등록 상표입니다.
©2024 Genentech, Inc. 모든 권리 보유.
환자 패키지 삽입물
| 이 환자 정보는 미국 식품의약국(FDA)의 승인을 받았습니다. | 개정: 2024년 04월 | ||
| 환자 정보 알렌센 (a-le-sen-sah) (알렉티닙) 캡슐 |
|||
| 알렌센에 대해 알아야 할 가장 중요한 정보는 무엇입니까? 알렌센은 다음과 같은 심각한 부작용을 일으킬 수 있습니다:
|
|||
|
|
||
“알렌센의 가능한 부작용은 무엇입니까?”를 참조하여 부작용에 대한 자세한 정보를 확인하십시오. |
|||
| 알렌센이란 무엇입니까? 알렌센은 비소세포폐암(NSCLC) 치료에 사용되는 처방약입니다. 이 비소세포폐암은 비정상적인 ananaplastic lymphoma kinase(ALK) 유전자로 인해 발생합니다:
의사는 알렌센이 귀하에게 적합한지 확인하기 위한 검사를 실시할 것입니다. |
|||
알렌센 복용 전, 다음과 같은 의학적 상태에 대해 의사에게 알리십시오:
복용 중인 모든 약물에 대해 의사에게 알리십시오, 처방약, 일반약, 비타민, 생약 보조제 등 |
|||
ALECENSA를 어떻게 복용해야 하나요?
|
|||
| ALECENSA 복용 중에 주의해야 할 사항은 무엇인가요? ALECENSA 치료 기간과 마지막 복용 후 7일 동안은 직사광선을 피하세요. 피부가 햇빛에 민감해질 수 있으며(광과민성) 쉽게 화상을 입고 심한 화상을 입을 수 있습니다. SPF 50 이상의 자외선 차단제와 립밤을 사용하는 등 화상 예방 조치를 취하세요. |
|||
| ALECENSA의 가능한 부작용은 무엇인가요? ALECENSA는 심각한 부작용을 유발할 수 있습니다. 다음을 포함합니다:
ALECENSA의 가장 흔한 부작용은 다음과 같습니다: |
|||
|
|
||
| 이것이 ALECENSA의 모든 가능한 부작용은 아닙니다. 자세한 정보는 의료 전문가나 약사에게 문의하세요. 부작용에 대한 의학적 조언이 필요하면 의사에게 연락하세요. FDA 1-800-FDA-1088번으로 부작용을 신고할 수 있습니다. |
|||
ALECENSA는 어떻게 보관해야 하나요?
ALECENSA와 모든 의약품을 어린이의 손이 닿지 않는 곳에 보관하세요. |
|||
| ALECENSA의 안전하고 효과적인 사용을 위한 일반 정보. 때때로 의약품은 환자 정보 안내문에 명시되지 않은 목적으로 처방됩니다. 처방되지 않은 상황에서는 ALECENSA를 사용하지 마세요. 증상이 비슷하더라도 ALECENSA를 다른 사람에게 주지 마세요. 해를 입힐 수 있습니다. 의료 전문가용 ALECENSA 정보는 약사나 의료 전문가에게 문의하세요. |
|||
| ALECENSA의 성분은 무엇인가요? 활성 성분: 알렉티닙 비활성 성분: 락토오스 일수화물, 하이드록시프로필 셀룰로오스, 나트륨 라우릴 설페이트, 스테아린산 마그네슘, 카르복시메틸셀룰로오스 칼슘. 캡슐 외피: 하이프로멜로오스, 캐러기난, 염화 칼륨, 이산화 타이타늄, 옥수수전분, 카르노버 왁스. 인쇄 잉크: 적색 산화철(E172), 황색 산화철(E172), FD&C 청색 제2호 알루미늄레이크(E132), 카르노버 왁스, 백셀락, 글리세릴 모노올레에이트. 판매원: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 ALECENSA®는 Chugai Pharmaceutical Co., Ltd., Tokyo, Japan의 등록상표입니다. ©2024 Genentech, Inc. 더 자세한 정보는 www.ALECENSA.com을 방문하거나 1-800-253-2367번으로 문의하세요. |
|||
주 디스플레이 패널 – 240캡슐 병 상자
NDC 50242-130-01
알렉센자®
(alectinib)
캡슐
150 mg
각 캡슐은 알렉티닙 150 mg
(알렉티닙 염산염 161.33 mg에 해당)을 함유합니다.
처방전 의약품
240캡슐
제넨텍
10166066
