
의약품 제조업체: Novartis Pharmaceuticals Corporation (Updated: 2024-07-23)
처방 정보의 주요 내용
PROMACTA® (엘트롬보파그) 정제, 경구용
PROMACTA® (엘트롬보파그) 경구용 현탁액
미국 최초 승인: 2008
적응증 및 사용
PROMACTA는 다음과 같은 경우에 사용되는 트롬보포이에틴 수용체 작용제입니다.
- 코르티코스테로이드, 면역 글로불린 또는 비장 절제술에 대한 반응이 불충분한 1세 이상의 성인 및 소아 환자의 지속적 또는 만성 면역 혈소판 감소증(ITP)의 혈소판 감소증 치료. PROMACTA는 혈소판 감소증의 정도와 임상 상태가 출혈 위험을 증가시키는 ITP 환자에게만 사용해야 합니다. (1.1)
- 인터페론 기반 치료의 시작 및 유지를 허용하기 위한 만성 C형 간염 환자의 혈소판 감소증 치료. PROMACTA는 혈소판 감소증의 정도가 인터페론 기반 치료의 시작을 방해하거나 인터페론 기반 치료의 유지를 제한하는 만성 C형 간염 환자에게만 사용해야 합니다. (1.2)
- 2세 이상의 성인 및 소아 환자의 중증 재생 불량성 빈혈의 1차 치료를 위한 표준 면역 억제 요법과 함께 사용. (1.3)
- 면역 억제 요법에 대한 반응이 불충분한 중증 재생 불량성 빈혈 환자의 치료. (1.3)
사용 제한:
투약 및 투여
- PROMACTA를 식사 없이 또는 칼슘 함량이 낮은 식사(≤ 50 mg)와 함께 복용하십시오. PROMACTA를 제산제, 칼슘이 풍부한 음식 및 미네랄 보충제와 같은 다가 양이온을 함유한 약물이나 제품을 복용하기 최소 2시간 전 또는 4시간 후에 복용하십시오. (2.4, 7.1, 12.3)
- 지속적 또는 만성 ITP: 대부분의 성인 및 6세 이상의 소아 환자에게는 PROMACTA를 1일 1회 50mg으로 시작하고, 1세에서 5세 사이의 소아 환자에게는 1일 1회 25mg으로 시작합니다. 간 기능 장애가 있는 환자와 동아시아/동남아시아계 환자 중 일부는 용량 감소가 필요합니다. 혈소판 수를 50 x 109/L 이상으로 유지하도록 조정합니다. 1일 75mg을 초과하지 마십시오. (2.1, 8.6, 8.7)
- 만성 C형 간염 관련 혈소판 감소증: 모든 환자에게 PROMACTA를 1일 1회 25mg으로 시작합니다. 항바이러스 요법을 시작하는 데 필요한 목표 혈소판 수를 달성하도록 조정합니다. 1일 용량을 100mg을 초과하지 마십시오. (2.2)
- 1차 중증 재생 불량성 빈혈: 표준 면역 억제 요법과 동시에 PROMACTA를 1일 1회 2.5 mg/kg(2세에서 5세 사이의 소아 환자), 75 mg(6세에서 11세 사이의 소아 환자) 또는 12세 이상의 환자에게는 150 mg으로 시작합니다. 동아시아/동남아시아계 환자는 초기 용량을 줄입니다. 독성 또는 혈소판 수 증가에 따라 용량을 조절합니다. (2.3, 8.7)
- 난치성 중증 재생 불량성 빈혈: PROMACTA를 1일 1회 50mg으로 시작합니다. 간 기능 장애가 있는 환자 또는 동아시아/동남아시아계 환자는 초기 용량을 줄입니다. 혈소판 수를 50 x 109/L 이상으로 유지하도록 조정합니다. 1일 150mg을 초과하지 마십시오. (2.3, 8.6, 8.7)
금기 사항
없음. (4)
경고 및 주의 사항
부작용
모든 적응증에서 가장 흔한 부작용(모든 적응증에서 ≥ 20%)은 빈혈, 메스꺼움, 발열, 알라닌 아미노트랜스퍼라제 증가, 기침, 피로, 두통 및 설사였습니다. (6.1)
의심되는 부작용을 보고하려면 Novartis Pharmaceuticals Corporation에 1-888-669-6682 또는 FDA에 1-800-FDA-1088로 연락하거나 www.fda.gov/medwatch를 방문하십시오.
특정 인구 집단에서의 사용
- 수유: 여성에게 치료 기간 동안 모유 수유를 하지 않도록 조언하십시오. (8.2)
환자 상담 정보 및 약물 안내는 17을 참조하십시오.
개정: 2023년 3월
목차
전문 정보: 목차*
경고: 만성 C형 간염 환자의 간 기능 저하 위험 및 간 독성 위험
1 적응증 및 사용법
1.1 지속적 또는 만성 면역 혈소판 감소증 환자의 혈소판 감소증 치료
1.2 C형 간염 감염 환자의 혈소판 감소증 치료
1.3 중증 재생불량성 빈혈 치료
1.4 사용 제한
2 용법 및 용량
2.1 지속적 또는 만성 면역 혈소판 감소증
2.2 만성 C형 간염 관련 혈소판 감소증
2.3 중증 재생불량성 빈혈
2.4 투여
3 제형 및 강도
4 금기 사항
5 경고 및 주의 사항
5.1 만성 C형 간염 환자의 간 기능 저하
5.2 간 독성
5.3 사망 위험 증가 및 골수이형성 증후군의 급성 골수성 백혈병으로의 진행 위험 증가
5.4 혈전증/색전증 합병증
5.5 백내장
6 이상 반응
6.1 임상 시험 경험
6.2 시판 후 경험
7 약물 상호 작용
7.1 다가 양이온 (킬레이션)
7.2 수송체
7.3 프로테아제 억제제
7.4 페그인터페론 알파-2a/b 치료
8 특정 환자군에서의 사용
8.1 임신
8.2 수유
8.3 생식 능력이 있는 여성 및 남성
8.4 소아 사용
8.5 노인 사용
8.6 간 기능 장애
8.7 민족
10 과량 투여
11 설명
12 약리학
12.1 작용 기전
12.2 약력학
12.3 약동학
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식 능력 저해
13.2 동물 약리학 및/또는 독성학
14 임상 연구
14.1 지속적 또는 만성 ITP
14.2 만성 C형 간염 관련 혈소판 감소증
14.3 중증 재생불량성 빈혈
16 포장 단위/보관 및 취급
16.1 정제
16.2 경구 현탁액용
17 환자 상담 정보
- *
- 전문 정보에서 생략된 섹션 또는 하위 섹션은 나열되지 않습니다.
제품에 대한 경고(BOXED WARNING)
경고: 만성 C형 간염 환자에서 간 기능 저하 위험 및 간독성 위험
만성 C형 간염 환자에서 인터페론 및 리바비린과 병용 투여 시 PROMACTA는 간 기능 저하 위험을 증가시킬 수 있습니다 [경고 및 주의 사항 (5.1) 참조].
PROMACTA는 심각하고 생명을 위협할 수 있는 간독성 위험을 증가시킬 수 있습니다. 권장 사항에 따라 간 기능을 모니터링하고 투약을 중단하십시오 [경고 및 주의 사항 (5.2) 참조].
1 적응증 및 용법
1.1 지속적 또는 만성 면역 혈소판 감소증 환자의 혈소판 감소증 치료
PROMACTA는 코르티코스테로이드, 면역 글로불린 또는 비장 절제술에 대한 반응이 불충분한 지속적 또는 만성 면역 혈소판 감소증(ITP)이 있는 1세 이상의 성인 및 소아 환자의 혈소판 감소증 치료에 사용됩니다. PROMACTA는 혈소판 감소증의 정도와 임상 상태가 출혈 위험을 증가시키는 ITP 환자에게만 사용해야 합니다.
1.2 C형 간염 감염 환자의 혈소판 감소증 치료
PROMACTA는 인터페론 기반 치료의 시작 및 유지가 가능하도록 만성 C형 간염 환자의 혈소판 감소증 치료에 사용됩니다. PROMACTA는 혈소판 감소증의 정도가 인터페론 기반 치료의 시작을 방해하거나 인터페론 기반 치료를 유지할 수 있는 능력을 제한하는 만성 C형 간염 환자에게만 사용해야 합니다.
1.3 중증 재생불량성 빈혈 치료
- PROMACTA는 2세 이상의 성인 및 소아 환자의 중증 재생불량성 빈혈의 1차 치료를 위한 표준 면역 억제 요법(IST)과 함께 사용됩니다.
- PROMACTA는 면역 억제 요법에 대한 반응이 불충분한 중증 재생불량성 빈혈 환자의 치료에 사용됩니다.
1.4 사용 제한
- PROMACTA는 골수이형성 증후군(MDS) 환자의 치료에 사용되지 않습니다. [경고 및 주의 사항(5.3) 참조].
- 만성 C형 간염 감염 치료를 위해 인터페론 없이 사용되는 직접 작용 항바이러스제와 병용한 경우 안전성 및 유효성이 확립되지 않았습니다.
2 투여 및 관리
2.1 지속적 또는 만성 면역 혈소판 감소증
출혈 위험을 줄이기 위해 필요에 따라 혈소판 수가 50 x 109/L 이상이 되도록 PROMACTA의 최저 용량을 사용하십시오. 용량 조절은 혈소판 수 반응을 기반으로 합니다. 혈소판 수를 정상화하기 위해 PROMACTA를 사용하지 마십시오 [경고 및 주의 사항 (5.4) 참조]. 임상 시험에서 혈소판 수는 일반적으로 PROMACTA를 시작한 후 1~2주 이내에 증가했고 PROMACTA를 중단한 후 1~2주 이내에 감소했습니다 [임상 연구 (14.1) 참조].
초기 용량 요법:
ITP가 있는 성인 및 6세 이상 소아 환자: 동아시아계 또는 경증에서 중증 간 기능 장애(Child-Pugh 등급 A, B, C)가 있는 환자를 제외하고 PROMACTA를 1일 1회 50mg으로 시작합니다.
ITP가 있는 동아시아계 환자의 경우 PROMACTA를 1일 1회 25mg의 감소된 용량으로 시작합니다 [특정 인구 집단에서의 사용 (8.7), 임상 약리학 (12.3) 참조].
ITP와 경증, 중등도 또는 중증 간 기능 장애(Child-Pugh 등급 A, B, C)가 있는 환자의 경우 PROMACTA를 1일 1회 25mg의 감소된 용량으로 시작합니다 [특정 인구 집단에서의 사용 (8.6), 임상 약리학 (12.3) 참조].
ITP와 간 기능 장애(Child-Pugh 등급 A, B, C)가 있는 동아시아계 환자의 경우 PROMACTA를 1일 1회 12.5mg의 감소된 용량으로 시작하는 것을 고려하십시오 [임상 약리학 (12.3) 참조].
ITP가 있는 1~5세 소아 환자: PROMACTA를 1일 1회 25mg으로 시작합니다 [특정 인구 집단에서의 사용 (8.7), 임상 약리학 (12.3) 참조].
모니터링 및 용량 조절: PROMACTA를 시작한 후 출혈 위험을 줄이기 위해 필요에 따라 혈소판 수가 50 x 109/L 이상이 되도록 용량을 조절합니다. 1일 75mg을 초과하지 마십시오. PROMACTA로 치료하는 동안 임상 혈액학 및 간 기능 검사를 정기적으로 모니터링하고 표 1에 요약된 대로 혈소판 수에 따라 PROMACTA의 용량 요법을 수정하십시오. PROMACTA로 치료하는 동안 혈소판 수를 포함한 차이를 포함한 완전 혈구 수(CBC)를 안정적인 혈소판 수가 달성될 때까지 매주 얻습니다. 그 후 혈소판 수를 포함한 차이를 포함한 CBC를 매월 얻습니다.
경구 현탁액과 정제를 전환할 때 2주 동안 매주 혈소판 수를 평가한 다음 표준 월별 모니터링을 따릅니다.
|
혈소판 수 결과 |
용량 조절 또는 반응 |
|
PROMACTA를 투여한 후 최소 2주 동안 < 50 x 109/L |
1일 용량을 25mg씩 증가시켜 최대 75mg/일까지 증가시킵니다. 1일 1회 12.5mg을 복용하는 환자의 경우 용량을 25mg으로 증가시킨 후 용량을 25mg씩 증가시킵니다. |
|
어느 때든 ≥ 200 x 109/L에서 ≤ 400 x 109/L |
1일 용량을 25mg씩 감소시킵니다. 이후 용량 조절의 효과를 평가하기 위해 2주를 기다립니다. 1일 1회 25mg을 복용하는 환자의 경우 용량을 1일 1회 12.5mg으로 감소시킵니다. |
|
> 400 x 109/L |
PROMACTA를 중단합니다. 혈소판 모니터링 빈도를 주 2회로 증가시킵니다. 혈소판 수가 < 150 x 109/L이 되면 1일 용량을 25mg 감소시켜 치료를 재개합니다. 1일 1회 25mg을 복용하는 환자의 경우 1일 12.5mg의 용량으로 치료를 재개합니다. |
|
PROMACTA의 최저 용량으로 치료를 시작한 후 2주 후 > 400 x 109/L |
PROMACTA를 중단합니다. |
ITP 및 간 기능 장애(Child-Pugh 등급 A, B, C) 환자의 경우 PROMACTA 투여 시작 후 또는 이후 용량 증가 후 용량 증가 전에 3주를 기다립니다.
PROMACTA 치료 중 혈소판 수치가 과도하게 증가하는 것을 방지하기 위해 의학적으로 적절한 경우 동반 ITP 약물의 용량 요법을 수정합니다. 24시간 내에 PROMACTA를 1회 이상 투여하지 마십시오.
중단: PROMACTA 최대 일일 용량 75mg으로 4주간 치료 후 혈소판 수치가 임상적으로 중요한 출혈을 피하기에 충분한 수준으로 증가하지 않으면 PROMACTA를 중단합니다. 표 1에 명시된 과도한 혈소판 수치 반응 또는 중요한 간 기능 검사 이상 또한 PROMACTA 중단을 필요로 합니다 [경고 및 주의 사항(5.2) 참조]. PROMACTA 중단 후 최소 4주 동안 매주 혈소판 수치를 포함한 차등 백혈구 수를 측정합니다.
2.2 만성 C형 간염 관련 혈소판 감소증
페길화 인터페론 및 리바비린으로 항바이러스 치료를 시작하고 유지하는 데 필요한 혈소판 수치를 달성하고 유지하기 위해 PROMACTA의 최저 용량을 사용합니다. 용량 조정은 혈소판 수치 반응을 기반으로 합니다. 혈소판 수치를 정상화하기 위해 PROMACTA를 사용하지 마십시오 [경고 및 주의 사항(5.4) 참조]. 임상 시험에서 혈소판 수치는 일반적으로 PROMACTA 치료 첫 주 안에 상승하기 시작했습니다 [임상 연구(14.2) 참조].
초기 용량 요법: PROMACTA를 1일 25mg으로 시작합니다.
모니터링 및 용량 조정: 항바이러스 치료를 시작하는 데 필요한 목표 혈소판 수치를 달성하기 위해 필요에 따라 2주마다 25mg씩 PROMACTA 용량을 조정합니다. 항바이러스 치료를 시작하기 전에 매주 혈소판 수치를 모니터링합니다.
항바이러스 치료 중에는 페길화 인터페론의 용량 감소를 피하기 위해 PROMACTA 용량을 조정합니다. 안정적인 혈소판 수치가 달성될 때까지 항바이러스 치료 중에 매주 차등 백혈구 수를 포함한 CBC를 모니터링합니다. 그 후 매월 혈소판 수치를 모니터링합니다. 1일 100mg을 초과하지 마십시오. PROMACTA 치료 전반에 걸쳐 임상 혈액학 및 간 기능 검사를 정기적으로 모니터링합니다.
페길화 인터페론 또는 리바비린에 대한 특정 용량 지침은 해당 처방 정보를 참조하십시오.
|
혈소판 수치 결과 |
용량 조정 또는 반응 |
|
PROMACTA 투여 후 최소 2주 동안 < 50 x 109/L |
일일 용량을 25mg 증가시켜 최대 100mg/일까지 증가시킵니다. |
|
어느 때든 ≥ 200 x 109/L에서 ≤ 400 x 109/L |
일일 용량을 25mg 감소시킵니다. 2주를 기다려 이러한 용량 조정 및 이후 용량 조정의 효과를 평가합니다. |
|
> 400 x 109/L |
PROMACTA를 중단합니다. 혈소판 모니터링 빈도를 주 2회로 증가시킵니다. 혈소판 수치가 < 150 x 109/L이 되면 25mg 감소된 일일 용량으로 치료를 재개합니다. 1일 25mg을 복용하는 환자의 경우 1일 12.5mg의 일일 용량으로 치료를 재개합니다. |
|
PROMACTA의 최저 용량으로 2주간 치료 후 > 400 x 109/L |
PROMACTA를 중단합니다. |
중단: 페길화 인터페론 및 리바비린의 처방 정보에는 치료 무효에 대한 항바이러스 치료 중단 권장 사항이 포함되어 있습니다. 치료 무효에 대한 항바이러스 치료 중단 권장 사항은 페길화 인터페론 및 리바비린 처방 정보를 참조하십시오.
항바이러스 치료를 중단하면 PROMACTA를 중단해야 합니다. 표 2에 명시된 과도한 혈소판 수치 반응 또는 중요한 간 기능 검사 이상 또한 PROMACTA 중단을 필요로 합니다 [경고 및 주의 사항(5.2) 참조].
2.3 중증 재생불량성 빈혈
1차 중증 재생불량성 빈혈
표준 면역 억제 요법과 동시에 PROMACTA를 시작합니다 [임상 연구(14.3) 참조].
초기 용량 요법
권장 초기 용량 요법은 표 3에 나와 있습니다. PROMACTA의 초기 용량을 초과하지 마십시오.
|
연령 |
용량 요법 |
|
12세 이상 환자 |
6개월 동안 1일 1회 150mg |
|
6세에서 11세까지의 소아 환자 |
6개월 동안 1일 1회 75mg |
|
2세에서 5세까지의 소아 환자 |
6개월 동안 1일 1회 2.5mg/kg |
동아시아/동남아시아계 혈통의 중증 재생불량성 빈혈 환자 또는 경증, 중등증 또는 중증 간 기능 장애(Child-Pugh 등급 A, B, C) 환자의 경우, 표 4에 나열된 것처럼 초기 PROMACTA 용량을 50% 감소시킵니다. [특정 인구 집단에서의 사용(8.6, 8.7), 임상 약리학(12.3) 참조].
기준 알라닌 아미노트랜스퍼라제(ALT) 또는 아스파르테이트 아미노트랜스퍼라제(AST) 수치가 정상 상한치(ULN)의 6배를 초과하는 경우, 트랜스아미나제 수치가 ULN의 5배 미만이 될 때까지 PROMACTA를 투여하지 마십시오. 이러한 환자의 경우 표 3 또는 표 4를 기반으로 초기 용량을 결정합니다.
|
연령 |
용량 요법 |
|
12세 이상 환자 |
6개월 동안 1일 1회 75mg |
|
6세에서 11세까지의 소아 환자 |
6개월 동안 1일 1회 37.5mg |
|
2세에서 5세까지의 소아 환자 |
6개월 동안 1일 1회 1.25mg/kg |
PROMACTA 모니터링 및 용량 조절: PROMACTA 치료 전반에 걸쳐 임상 혈액학 및 간 기능 검사를 정기적으로 실시합니다. [경고 및 주의 사항(5.2) 참조].
표 5에 요약된 것처럼 혈소판 수치에 따라 PROMACTA 용량 요법을 수정합니다.
|
혈소판 수치 결과 |
용량 조절 또는 반응 |
|
> 200 x 109/L에서 ≤ 400 x 109/L |
2주마다 25mg씩 매일 용량을 감소시켜 혈소판 수치가 ≥ 50 x 109/L를 유지하는 최저 용량으로 감소시킵니다. |
|
> 400 x 109/L |
PROMACTA를 1주일 동안 중단합니다. 혈소판 수치가 < 200 x 109/L가 되면 25mg(또는 12세 미만의 소아 환자의 경우 12.5mg) 감소된 매일 용량으로 PROMACTA를 재투여합니다. |
표 6은 상승된 간 트랜스아미나제 수치 및 혈전색전증 사건 관리에서 PROMACTA 용량 중단, 감소 또는 중단에 대한 권장 사항을 요약합니다.
| 약어: ALT, 알라닌 아미노트랜스퍼라제; AST, 아스파르테이트 아미노트랜스퍼라제; ULN, 정상 상한치. | |
|
사건 |
권장 사항 |
|
ALT 또는 AST 상승 |
ALT 또는 AST 상승 > 6 x ULN
PROMACTA 재투여 후 ALT 또는 AST 상승 > 6 x ULN
감소된 용량으로 ALT 또는 AST가 다시 > 6 x ULN으로 돌아오는 경우 12세 미만의 소아 환자의 경우 매일 용량을 최소 15% 감소시켜 투여 가능한 가장 가까운 용량으로 감소시킵니다. |
|
혈전색전증 사건(예: 심부 정맥 혈전증, 폐색전증, 뇌졸중, 심근 경색) |
PROMACTA를 중단하지만 말 항림프구 글로불린(h-ATG) 및 사이클로스포린은 계속 투여합니다. |
PROMACTA 치료의 총 기간은 6개월입니다.
난치성 중증 재생불량성 빈혈
혈액학적 반응을 얻고 유지하기 위해 PROMACTA의 최저 용량을 사용하십시오. 용량 조절은 혈소판 수를 기반으로 합니다. 혈액학적 반응에는 용량 적정이 필요하며, 일반적으로 최대 150mg까지 적정하며 PROMACTA를 시작한 후 최대 16주까지 걸릴 수 있습니다 [임상 연구(14.3) 참조].
초기 용량 요법: PROMACTA를 1일 50mg으로 시작하십시오.
동아시아/동남아시아계 중증 재생불량성 빈혈 환자 또는 경증, 중등도 또는 중증 간 기능 장애(Child-Pugh 등급 A, B, C) 환자의 경우 PROMACTA를 1일 25mg의 감소된 용량으로 시작하십시오 [특정 인구 집단에서의 사용(8.6, 8.7), 임상 약리학(12.3) 참조].
모니터링 및 용량 조절: 필요에 따라 혈소판 수가 50 x 109/L 이상이 되도록 2주마다 50mg씩 PROMACTA 용량을 조절하십시오. 1일 150mg을 초과하지 마십시오. PROMACTA 치료 전반에 걸쳐 임상 혈액학 및 간 기능 검사를 정기적으로 모니터링하고 표 7에 요약된 대로 혈소판 수에 따라 PROMACTA의 용량 요법을 수정하십시오.
|
혈소판 수 결과 |
용량 조절 또는 반응 |
|
PROMACTA를 투여한 후 최소 2주 동안 < 50 x 109/L |
1일 용량을 50mg씩 증가시켜 최대 150mg/일까지 증가시킵니다. 1일 25mg을 복용하는 환자의 경우 용량을 50mg으로 증가시킨 후 용량을 50mg씩 증가시킵니다. |
|
언제든지 ≥ 200 x 109/L에서 ≤ 400 x 109/L |
1일 용량을 50mg씩 감소시킵니다. 이후 용량 조절의 효과를 평가하기 위해 2주를 기다립니다. |
|
> 400 x 109/L |
PROMACTA를 1주일 동안 중단합니다. 혈소판 수가 < 150 x 109/L이 되면 50mg 감소된 용량으로 치료를 재개합니다. |
|
PROMACTA의 최저 용량으로 2주간 치료 후 > 400 x 109/L |
PROMACTA를 중단합니다. |
최소 8주 동안 지속되는 수혈 독립성을 포함한 삼계열 반응을 보이는 환자의 경우 PROMACTA 용량을 50% 감소시킬 수 있습니다 [임상 연구(14.3) 참조]. 감소된 용량으로 8주 후에 수치가 안정적으로 유지되면 PROMACTA를 중단하고 혈액 수치를 모니터링합니다. 혈소판 수가 30 x 109/L 미만, 헤모글로빈이 9g/dL 미만 또는 절대 호중구 수(ANC)가 0.5 x 109/L 미만으로 떨어지면 이전에 효과적인 용량으로 PROMACTA를 재개할 수 있습니다.
중단: PROMACTA로 16주간 치료를 받은 후 혈액학적 반응이 나타나지 않으면 치료를 중단하십시오. 새로운 세포 유전학적 이상이 관찰되면 PROMACTA 중단을 고려하십시오 [부작용(6.1) 참조]. 표 7에 요약된 과도한 혈소판 수 반응 또는 중요한 간 기능 검사 이상도 PROMACTA 중단을 필요로 합니다 [경고 및 주의 사항(5.2) 참조].
2.4 투여
정제 및 구강 현탁액 투여: PROMACTA를 식사 없이 또는 칼슘 함량이 낮은 식사(≤ 50mg)와 함께 복용하십시오. PROMACTA를 다른 약물(예: 제산제), 칼슘이 풍부한 식품(50mg 이상의 칼슘 함유, 예: 유제품, 칼슘 강화 주스 및 특정 과일과 채소), 또는 철, 칼슘, 알루미늄, 마그네슘, 셀레늄 및 아연과 같은 다가 양이온을 함유한 보충제를 복용하기 최소 2시간 전 또는 4시간 후에 복용하십시오 [약물 상호 작용(7.1), 임상 약리학(12.3) 참조].
정제를 쪼개거나 씹거나 부수지 말고 음식이나 액체와 섞지 마십시오.
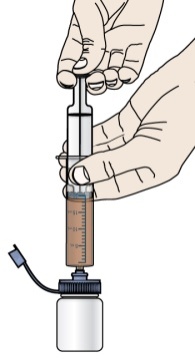
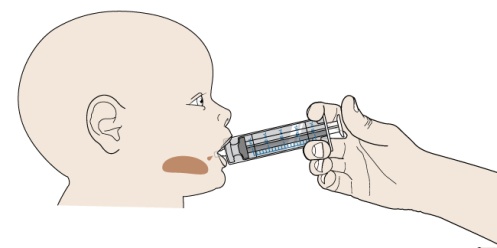
구강 현탁액 제조: 구강 현탁액을 사용하기 전에 환자 또는 보호자가 구강 현탁액용 PROMACTA의 적절한 용량, 제조 및 투여에 대한 교육을 받았는지 확인하십시오.
제조 후 즉시 구강 현탁액을 투여하십시오. 제조 후 30분 이내에 투여하지 않은 현탁액은 버리십시오.
물만 사용하여 현탁액을 제조하십시오. 참고: 현탁액을 제조할 때 뜨거운 물을 사용하지 마십시오.
각 구강 용량 주사기의 권장 사용 기간을 포함하여 현탁액의 제조 및 투여에 대한 자세한 내용은 [사용 지침]을 참조하십시오.
3 제형 및 함량
정제
- 12.5mg 정제 —– 둥글고 양쪽으로 볼록하며, 흰색의 필름 코팅 정제로 한쪽 면에 “GS MZ1”과 12.5가 각인되어 있습니다. 경구 투여용 각 정제는 엘트롬보파그 유리산 12.5mg에 해당하는 엘트롬보파그 올라민을 함유하고 있습니다.
- 25mg 정제 —– 둥글고 양쪽으로 볼록하며, 주황색의 필름 코팅 정제로 한쪽 면에 “GS NX3”과 25가 각인되어 있습니다. 경구 투여용 각 정제는 엘트롬보파그 유리산 25mg에 해당하는 엘트롬보파그 올라민을 함유하고 있습니다.
- 50mg 정제 —– 둥글고 양쪽으로 볼록하며, 파란색의 필름 코팅 정제로 한쪽 면에 “GS UFU”와 50이 각인되어 있습니다. 경구 투여용 각 정제는 엘트롬보파그 유리산 50mg에 해당하는 엘트롬보파그 올라민을 함유하고 있습니다.
- 75mg 정제 —– 둥글고 양쪽으로 볼록하며, 분홍색의 필름 코팅 정제로 한쪽 면에 “GS FFS”와 75가 각인되어 있습니다. 경구 투여용 각 정제는 엘트롬보파그 유리산 75mg에 해당하는 엘트롬보파그 올라민을 함유하고 있습니다.
경구 현탁액용
- 12.5mg 포 —– 재구성을 위한 적갈색에서 노란색 분말을 함유하고 있습니다.
- 25mg 포 —– 재구성을 위한 적갈색에서 노란색 분말을 함유하고 있습니다.
4 금기사항
없음.
5 경고 및 주의사항
5.1 만성 C형 간염 환자의 간 기능 저하
만성 C형 간염 환자에서 PROMACTA를 인터페론 및 리바비린과 병용하면 간 기능 저하 위험이 증가할 수 있습니다. 만성 C형 간염 및 혈소판 감소증 환자를 대상으로 한 두 건의 대조군 임상 시험에서, 복수 및 뇌병증은 PROMACTA와 항바이러스제를 병용 투여한 군(7%)에서 위약과 항바이러스제를 병용 투여한 군(4%)보다 더 빈번하게 발생했습니다. 기저 시점에 알부민 수치가 낮은(3.5g/dL 미만) 환자 또는 말기 간 질환 모델(MELD) 점수가 10 이상인 환자는 PROMACTA와 항바이러스제를 병용 투여한 군에서 간 기능 저하 위험이 더 높았습니다. 항바이러스 요법을 중단하면 PROMACTA를 중단하십시오.
5.2 간 독성
PROMACTA는 심각하고 생명을 위협할 수 있는 간 독성 위험을 증가시킬 수 있습니다. [유해 반응(6.1) 참조]. 임상 시험에서 PROMACTA로 치료받은 ITP 환자 1명(< 1%)에서 약물 유발성 간 손상이 발생했습니다. 임상 시험에서 PROMACTA로 치료받은 만성 C형 간염 환자 11명(1%)에서 약물 유발성 간 손상이 발생했습니다.
ITP, 만성 C형 간염 관련 혈소판 감소증 및 난치성 중증 재생불량성 빈혈 치료
PROMACTA 투여 시작 전, 용량 조절 단계 동안 2주마다, 안정적인 용량 확립 후 매월 혈청 ALT, AST 및 빌리루빈을 측정하십시오. PROMACTA는 UDP-글루쿠로노실 전이효소(UGT)1A1 및 유기 음이온 수송 폴리펩티드(OATP)1B1을 억제하여 간접적 고빌리루빈혈증을 유발할 수 있습니다. 빌리루빈이 상승하면 분획화를 수행하십시오. 3~5일 이내에 반복 검사를 통해 비정상적인 혈청 간 기능 검사를 평가하십시오. 이상이 확인되면 혈청 간 기능 검사를 해결되거나 안정될 때까지 매주 모니터링하십시오. 정상적인 간 기능을 가진 환자의 경우 ALT 수치가 ULN의 3배 이상이거나, 전처치 시 트랜스아미나제 수치가 상승한 환자의 경우 기저 수치의 3배 이상(또는 ULN의 5배 이상, 둘 중 낮은 값)으로 증가하고 다음과 같은 경우 PROMACTA를 중단하십시오.
- 지속적으로 증가하거나,
- 4주 이상 지속되거나,
- 직접 빌리루빈 증가를 동반하거나,
- 간 손상의 임상 증상 또는 간 기능 저하 증거를 동반합니다.
PROMACTA로 재투여 치료의 잠재적 이점이 간 독성 위험보다 크다고 판단되는 경우, PROMACTA를 신중하게 재투여하고 용량 조절 단계 동안 매주 혈청 간 기능 검사를 측정하십시오. PROMACTA를 재투여하면 간 독성이 재발할 수 있습니다. 간 기능 검사 이상이 지속되거나 악화되거나 재발하면 PROMACTA를 영구적으로 중단하십시오.
중증 재생불량성 빈혈의 1차 치료
PROMACTA 투여 시작 전, h-ATG 요법으로 입원 중 매일, 치료 중 2주마다 혈청 ALT, AST 및 빌리루빈을 측정하십시오. 치료 중 ALT 또는 AST 수치 증가는 표 6에 권장된 대로 관리하십시오.
5.3 사망 위험 증가 및 골수이형성 증후군의 급성 골수성 백혈병으로의 진행 위험 증가
국제 예후 점수 시스템(IPSS) 중간 1, 중간 2 또는 고위험 MDS 및 혈소판 감소증 환자를 대상으로 한 무작위 배정, 이중 맹검, 위약 대조군, 다기관 임상 시험에서, PROMACTA(n = 179) 또는 위약(n = 177)과 병용하여 아자시티딘을 투여받은 환자는 효능 부족 및 안전성 문제(급성 골수성 백혈병(AML)으로의 진행 증가 포함)로 인해 중단되었습니다. 환자는 아자시티딘과 병용하여 최대 1일 300mg까지 1일 1회 200mg의 시작 용량으로 PROMACTA 또는 위약을 최소 6주기 동안 투여받았습니다. 사망률(전반적인 생존율)은 PROMACTA 군에서 32%(57/179)였고 위약 군에서 29%(51/177)였습니다(HR [95% CI] = 1.42 [0.97, 2.08], 이 임상 시험에서 PROMACTA 군의 사망 위험이 42% 증가한 것으로 나타났습니다). AML로의 진행률은 PROMACTA 군에서 12%(21/179)였고 위약 군에서 6%(10/177)였습니다(HR [95% CI] = 2.66 [1.31, 5.41], 이 임상 시험에서 PROMACTA 군의 AML로의 진행 위험이 166% 증가한 것으로 나타났습니다).
5.4 혈전증/색전증 합병증
PROMACTA로 혈소판 수치가 증가하면 혈전증/색전증 합병증이 발생할 수 있습니다. 보고된 혈전증/색전증 합병증에는 정맥 및 동맥 사건이 모두 포함되었으며, 혈소판 수치가 낮거나 정상인 경우에 관찰되었습니다.
혈전색전증 위험 요인이 있는 환자(예: Factor V Leiden, ATIII 결핍, 항인지질 증후군, 만성 간 질환)에게 PROMACTA를 투여할 때 혈전색전증 위험이 증가할 가능성을 고려하십시오. 혈전증/색전증 합병증 위험을 최소화하기 위해, PROMACTA를 사용하여 혈소판 수치를 정상화하려고 하지 마십시오. [용법 및 용량(2.1, 2.2, 2.3) 참조]에 따라 용량 조절 지침을 따르십시오. 목표 혈소판 수치를 달성하고 유지하십시오.
만성 C형 간염 및 혈소판 감소증 환자를 대상으로 한 두 건의 대조군 임상 시험에서, PROMACTA로 치료받은 환자의 3%(31/955)에서 혈전증 사건이 발생한 반면, 위약 투여 환자의 1%(5/484)에서 혈전증 사건이 발생했습니다. 대부분의 사건은 문맥계(PROMACTA로 치료받은 환자의 1% 대 위약 투여 환자의 1% 미만)였습니다.
만성 간 질환 및 ITP와 관련 없는 혈소판 감소증 환자를 대상으로 한 통제된 임상 시험에서 선택적 침습적 절차를 받은 환자(N = 292)의 경우, PROMACTA 75mg을 1일 1회 투여한 환자에서 혈전증 발생 위험이 증가했습니다. PROMACTA를 투여받은 그룹에서 7건의 혈전증 합병증(6명의 환자)이 보고되었고, 위약 그룹에서 3건의 혈전증 합병증(2명의 환자)이 보고되었습니다. PROMACTA를 투여받은 그룹에서 보고된 모든 혈전증 합병증은 문맥 혈전증(PVT)이었습니다. PVT의 증상으로는 복통, 메스꺼움, 구토 및 설사가 포함되었습니다. PROMACTA를 투여받은 그룹의 6명 환자 중 5명은 PROMACTA 치료를 완료한 후 30일 이내에 혈소판 수가 200 x 109/L를 초과한 상태에서 혈전증 합병증을 경험했습니다. 침습적 절차를 위해 2주 동안 PROMACTA 75mg을 1일 1회 투여한 만성 간 질환 환자의 혈소판 감소증 환자에서 문맥 혈전증 위험이 증가했습니다.
5.5 백내장
지속적 또는 만성 ITP 성인을 대상으로 한 3건의 통제된 임상 시험에서 PROMACTA 50mg을 매일 투여받은 환자 15명(7%)과 위약 그룹 환자 8명(7%)에서 백내장이 발생하거나 악화되었습니다. 연장 시험에서 PROMACTA 치료 전 안과 검사를 받은 환자의 11%에서 백내장이 발생하거나 악화되었습니다. 만성 C형 간염 및 혈소판 감소증 환자를 대상으로 한 2건의 통제된 임상 시험에서 PROMACTA로 치료받은 환자의 8%와 위약으로 치료받은 환자의 5%에서 백내장이 발생하거나 악화되었습니다.
백내장은 설치류에서 엘트롬보파그의 독성 연구에서 관찰되었습니다 [비임상 독성학(13.2) 참조]. PROMACTA 투여 전에 기준 안과 검사를 실시하고 PROMACTA 치료 중에 백내장의 징후와 증상을 정기적으로 모니터링하십시오.
6 부작용
다음 임상적으로 중요한 PROMACTA 관련 이상 반응은 다른 섹션에 설명되어 있습니다.
- 만성 C형 간염 환자의 간 기능 저하 [경고 및 주의 사항 (5.1) 참조]
- 간 독성 [경고 및 주의 사항 (5.2) 참조]
- 사망 위험 증가 및 골수 이형성 증후군의 급성 골수성 백혈병으로의 진행 [경고 및 주의 사항 (5.3) 참조]
- 혈전증/색전증 합병증 [경고 및 주의 사항 (5.4) 참조]
- 백내장 [경고 및 주의 사항 (5.5) 참조]
6.1 임상 시험 경험
임상 시험은 매우 다양한 조건에서 수행되므로 약물의 임상 시험에서 관찰된 이상 반응 발생률을 다른 약물의 임상 시험에서 관찰된 발생률과 직접 비교할 수 없으며 실제로 관찰된 발생률을 반영하지 않을 수 있습니다.
지속적 또는 만성 면역 혈소판 감소증
성인: 임상 시험에서 출혈은 가장 흔한 중대한 이상 반응이었으며 대부분의 출혈 반응은 PROMACTA 중단 후 발생했습니다. 다른 중대한 이상 반응으로는 혈전증/색전증 합병증 [경고 및 주의 사항 (5.4) 참조]이 포함되었습니다. 아래에 설명된 데이터는 3개의 위약 대조 시험과 1개의 공개 표지 확장 시험 [임상 연구 (14.1) 참조]에서 18세에서 85세 사이의 지속적 또는 만성 ITP 환자(여성 66%)에 대한 PROMACTA 노출을 반영합니다. PROMACTA는 330명의 환자에게 최소 6개월 동안, 218명의 환자에게 최소 1년 동안 투여되었습니다.
표 8은 3개의 위약 대조 시험에서 PROMACTA를 투여받은 환자의 3% 이상에서 발생한 가장 흔한 이상 약물 반응(PROMACTA 대 위약에서 발생률이 더 높음)을 보여줍니다.
| 약어: ALT, 알라닌 아미노 전이효소; AST, 아스파르트산 아미노 전이효소. a요로 감염, 방광염, 세균성 요로 감염 및 세균뇨를 포함합니다. |
||
|
이상 반응 |
PROMACTA 50 mg n = 241 (%) |
위약 n = 128 (%) |
|
메스꺼움 |
9 |
3 |
|
설사 |
9 |
7 |
|
상기도 감염 |
7 |
6 |
|
구토 |
6 |
< 1 |
|
요로 감염a |
5 |
4 |
|
ALT 증가 |
5 |
3 |
|
근육통 |
5 |
2 |
|
구강 인두 통증 |
4 |
3 |
|
AST 증가 |
4 |
2 |
|
인후염 |
4 |
2 |
|
요통 |
3 |
2 |
|
독감 |
3 |
2 |
|
감각 이상 |
3 |
2 |
|
발진 |
3 |
2 |
3개의 통제된 임상 지속성 또는 만성 ITP 시험에서 탈모, 근골격계 통증, 혈액 알칼리성 포스파타제 증가 및 구강 건조는 PROMACTA로 치료받은 환자의 2%에서 보고되었으며 위약을 투여받은 환자에게는 보고되지 않았습니다.
단일군 연장 시험에서 PROMACTA를 투여받은 지속성 또는 만성 ITP 환자 302명 중 부작용은 위약 대조 시험에서 관찰된 패턴과 유사하게 발생했습니다. 표 9는 연장 시험에서 PROMACTA를 투여받은 환자의 3% 이상에서 발생한 가장 흔한 치료 관련 부작용을 보여줍니다.
| 약어: ALT, 알라닌 아미노 전이효소; AST, 아스파르트산 아미노 전이효소. | |
|
부작용 |
PROMACTA 50 mg n = 302 (%) |
|
두통 |
10 |
|
ALT 증가 |
5 |
|
AST 증가 |
5 |
|
백내장 |
5 |
|
피로 |
5 |
|
혈액 빌리루빈 증가 |
4 |
|
메스꺼움 |
4 |
|
고빌리루빈혈증 |
3 |
|
설사 |
3 |
3개의 통제된 지속적 또는 만성 ITP 임상시험에서, PROMACTA 투여군의 경우 혈청 간 기능 검사 이상(주로 중증도 2등급 이하)이 각각 11%와 7%로 보고되었으며, 위약 투여군의 경우 각각 11%와 7%로 보고되었습니다. PROMACTA 투여군의 경우 4명(1%)의 환자가, 위약 투여군의 경우 3명(2%)의 환자가 간담도계 실험실 검사 이상으로 인해 치료를 중단했습니다. 간담도계 실험실 검사 이상이 나타난 통제된 임상시험에서 PROMACTA 투여군의 환자 17명은 연장 임상시험에서 PROMACTA에 재노출되었습니다. 이들 환자 중 8명은 다시 간 기능 검사 이상(3등급 이하)을 경험했으며, 그 결과 1명의 환자가 PROMACTA 투여를 중단했습니다. 연장 지속적 또는 만성 ITP 임상시험에서 6명의 환자가 추가로 간 기능 검사 이상(3등급 이하)으로 인해 PROMACTA 투여를 중단했습니다.
3개의 통제된 지속적 또는 만성 ITP 임상시험에서, PROMACTA 투여군의 경우 7%의 환자가 백내장이 발생하거나 악화되었으며, 위약 투여군의 경우 7%의 환자가 백내장이 발생하거나 악화되었습니다. 모든 환자는 코르티코스테로이드 사용을 포함한 백내장 발생 위험 요인이 사전에 문서화되었습니다. 연장 임상시험에서, PROMACTA 치료 전 안과 검사를 받은 환자의 11%에서 백내장이 발생하거나 악화되었습니다. 환자의 72%는 코르티코스테로이드 사용을 포함한 사전에 존재하는 위험 요인이 있었습니다.
PROMACTA의 안전성은 7개의 성인 지속적 또는 만성 ITP 임상시험(PROMACTA 투여군 763명, 위약 투여군 179명)에서 치료받은 모든 환자를 대상으로 평가되었습니다. PROMACTA 투여군의 경우 6%의 환자에서 혈전색전증이 보고되었으며, 위약 투여군의 경우 0%의 환자에서 혈전색전증이 보고되었습니다. PROMACTA 투여군의 경우 1% 미만의 환자에서 급성 신부전을 동반한 혈전성 미세혈관병증이 보고되었으며, 위약 투여군의 경우 0%의 환자에서 혈전성 미세혈관병증이 보고되었습니다.
ITP와 관련되지 않은 만성 간 질환 및 혈소판 감소증 환자를 대상으로 한 PROMACTA의 위약 대조 임상시험에서, PROMACTA 투여군의 경우 6명의 환자가, 위약 투여군의 경우 1명의 환자가 문맥 정맥 혈전증을 발생했습니다. [경고 및 주의 사항(5.4) 참조]
소아 환자: 아래에 설명된 데이터는 2개의 위약 대조 임상시험의 무작위 배정 단계에서 지속적 또는 만성 ITP를 가진 소아 환자(1세에서 17세까지, 107명)에 대한 PROMACTA의 중간 노출 기간(91일)을 반영하며, 이 중 53%가 여성이었습니다.
표 10은 2개의 위약 대조 임상시험에서 PROMACTA 투여군과 위약 투여군 간에 발생 빈도가 더 높은 가장 흔한 약물 유해 반응(PROMACTA를 투여받은 1세 이상의 소아 환자의 3% 이상에서 발생)을 보여줍니다.
| 약어: ALT, 알라닌 아미노 전이효소; AST, 아스파르트산 아미노 전이효소. aULN의 3배를 초과하는 약물 유해 반응 또는 실험실 검사 이상을 포함합니다. |
||
|
PROMACTA |
위약 |
|
|
n = 107 |
n = 50 |
|
|
약물 유해 반응 |
(%) |
(%) |
|
상기도 감염 |
17 |
6 |
|
비인두염 |
12 |
4 |
|
기침 |
9 |
0 |
|
설사 |
9 |
2 |
|
발열 |
9 |
8 |
|
복통 |
8 |
4 |
|
구강인두 통증 |
8 |
2 |
|
치통 |
6 |
0 |
|
ALT 증가a |
6 |
0 |
|
발진 |
5 |
2 |
|
AST 증가 |
4 |
0 |
|
비염 |
4 |
0 |
두 건의 대조군 임상 시험에서 지속적 또는 만성 ITP 환자 중 PROMACTA로 치료받은 환자 2명(1%)에게서 백내장이 발생하거나 악화되었습니다. 두 환자 모두 백내장 발생 위험 요인인 만성 경구 코르티코스테로이드를 투여받았습니다.
만성 C형 간염 관련 혈소판 감소증: 두 건의 위약 대조군 임상 시험에서 만성 C형 간염 관련 혈소판 감소증 환자 955명이 PROMACTA를 투여받았습니다. 표 11은 PROMACTA를 투여받은 환자 중 위약군과 비교하여 10% 이상에서 발생한 가장 흔한 약물 이상 반응을 보여줍니다.
| a불면증, 초기 불면증 및 수면 질 저하의 PT를 포함합니다. | ||
|
약물 이상 반응 |
PROMACTA + 페길 인터페론/리바비린 n = 955 (%) |
위약 + 페길 인터페론/리바비린 n = 484 (%) |
|
빈혈 |
40 |
35 |
|
발열 |
30 |
24 |
|
피로 |
28 |
23 |
|
두통 |
21 |
20 |
|
메스꺼움 |
19 |
14 |
|
설사 |
19 |
11 |
|
식욕 감소 |
18 |
14 |
|
독감 유사 증상 |
18 |
16 |
|
불면증a |
16 |
15 |
|
쇠약 |
16 |
13 |
|
기침 |
15 |
12 |
|
가려움증 |
15 |
13 |
|
오한 |
14 |
9 |
|
근육통 |
12 |
10 |
|
탈모 |
10 |
6 |
|
말초 부종 |
10 |
5 |
발진은 PROMACTA를 투여받은 환자의 9%와 위약을 투여받은 환자의 7%에서 보고되었습니다.
만성 C형 간염 환자를 대상으로 한 두 건의 대조군 임상 시험에서, PROMACTA를 투여받은 환자의 8%에서 위약 투여 환자의 3%에 비해 고빌리루빈혈증이 보고되었습니다. 총 빌리루빈이 ULN의 1.5배 이상인 경우는 PROMACTA를 투여받은 환자의 76%와 위약을 투여받은 환자의 50%에서 보고되었습니다. ALT 또는 AST가 ULN의 3배 이상인 경우는 PROMACTA를 투여받은 환자의 34%와 위약을 투여받은 환자의 38%에서 보고되었습니다.
만성 C형 간염 환자를 대상으로 한 두 건의 대조군 임상 시험에서, PROMACTA로 치료받은 환자의 8%와 위약으로 치료받은 환자의 5%에서 백내장이 발생하거나 악화되었습니다.
PROMACTA의 안전성은 두 건의 대조군 임상 시험에서 PROMACTA로 치료받은 모든 환자(시험의 항바이러스 치료 전 단계에서 처음으로 PROMACTA를 투여받고 나중에 위약군에 무작위 배정된 환자 포함, N = 1520명의 PROMACTA 투여 환자)를 대상으로 평가되었습니다. 간부전은 PROMACTA 투여 환자의 0.8%와 위약 투여 환자의 0.4%에서 보고되었습니다.
중증 재생불량성 빈혈
중증 재생불량성 빈혈의 1차 치료
PROMACTA의 안전성은 이전에 확실한 면역억제 요법을 받지 않은 중증 재생불량성 빈혈 환자 153명을 대상으로 한 단일군 시험을 기반으로 확립되었습니다. 이 시험에서 PROMACTA는 말 항흉선 글로불린(h-ATG)과 사이클로스포린과 병용하여 투여되었습니다. [임상 연구(14.3) 참조] 이 시험에서 투약을 받은 153명의 환자 중 92명은 권장 용량 및 일정으로 PROMACTA, h-ATG 및 사이클로스포린을 병용 사용한 안전성을 평가할 수 있었습니다.
이 코호트에서 PROMACTA는 1일 1회 최대 150mg을 1일부터 6개월까지(D1-M6) 투여하고, h-ATG는 1일부터 4일까지 투여하고, 사이클로스포린은 6개월 동안 투여했습니다. 그 후 6개월 만에 혈액학적 반응을 보인 환자는 추가로 18개월 동안 저용량 사이클로스포린(유지 용량)을 투여했습니다. 이 코호트에서 PROMACTA에 대한 중간 노출 기간은 183일이었으며, 환자의 70%가 24주 이상 노출되었습니다.
표 12는 D1-M6 코호트에서 PROMACTA와 관련된 가장 흔한 부작용(환자의 5% 이상에서 발생)을 보여줍니다.
| 약어: ALT, 알라닌 아미노트랜스퍼라제; AST, 아스파르테이트 아미노트랜스퍼라제. | |
|
부작용 |
PROMACTA |
|
ALT 증가 |
29 |
|
AST 증가 |
17 |
|
혈액 빌리루빈 증가 |
17 |
|
발진 |
8 |
|
피부 변색, 과색소침착 포함 |
5 |
PROMACTA D1-M6 코호트에서 ALT 증가(29%), AST 증가(17%), 혈액 빌리루빈 증가(17%)가 난치성 중증 재생불량성 빈혈 환자보다 더 빈번하게 보고되었습니다(표 13 참조).
PROMACTA D1-M6 코호트에서 새로운 또는 악화된 간 기능 검사 이상(CTCAE 등급 3 및 등급 4)은 AST의 경우 각각 15% 및 2%, ALT의 경우 26% 및 4%, 빌리루빈의 경우 12% 및 1%였습니다.
이 단일군 오픈 라벨 임상 시험에서 PROMACTA D1-M6 코호트에서 총 빌리루빈 > 1.5 x ULN인 ALT 또는 AST > 3 x ULN 및 총 빌리루빈 > 2 x ULN인 ALT 또는 AST > 3 x ULN이 각각 44% 및 32%의 환자에서 보고되었습니다.
소아 환자
총 34명의 소아 환자(2세에서 5세까지 2명, 6세에서 11세까지 12명, 12세에서 16세까지 20명)가 이 단일군 시험에 등록되었으며, 그 중 26명의 소아 환자가 PROMACTA D1-M6 코호트에 등록되었습니다. 이 코호트에서 가장 빈번한 중대한 유해 반응(환자의 ≥ 10%에서 발생)은 상기도 감염(2세에서 16세까지 환자의 12% 대 17세 이상 환자의 5%)과 발진(12% 대 2%)이었습니다. PROMACTA와 관련된 가장 흔한 유해 반응(환자의 ≥ 10%에서 발생)은 ALT 증가(2세에서 16세까지 환자의 23% 대 17세 이상 환자의 32%), 혈액 빌리루빈 증가(12% 대 20%), AST 증가(12% 대 20%), 발진(12% 대 6%)이었습니다.
세포 유전학적 이상
이 시험에서 환자는 세포 유전학적 이상에 대해 골수 천자를 평가받았습니다. PROMACTA D1-M6 코호트에서 7명의 환자에게 새로운 세포 유전학적 이상이 보고되었으며, 그 중 4명은 7번 염색체 손실이 있었습니다. 이 4명은 6.1개월 이내에 발생했습니다. 모든 코호트에서 153명 중 15명(10%)의 환자에서 클론 세포 유전학적 진화가 발생했습니다. 세포 유전학적 이상을 경험한 15명의 환자 중 7명은 7번 염색체 손실이 있었고, 그 중 6명은 6.1개월 이내에 발생했습니다. 4명의 환자는 의미가 불분명한 염색체 이상이 있었습니다. 3명의 환자는 13번 염색체 결손이 있었습니다. 1명의 환자는 5년 후 골수 평가를 받았는데, 이때 골수 이형성 증후군의 잠재적 발병과 관련하여 과형성을 동반한 이형성의 특징이 나타났습니다. 이러한 발견이 기저 질환, 면역 억제 요법, 또는 PROMACTA 치료로 인해 발생했는지 여부는 불분명합니다.
난치성 중증 재생불량성 빈혈
단일군 오픈 라벨 시험에서 난치성 중증 재생불량성 빈혈이 있는 43명의 환자가 PROMACTA를 투여받았습니다. 11명의 환자(26%)는 6개월 이상 치료를 받았고, 7명의 환자(16%)는 1년 이상 치료를 받았습니다. 가장 흔한 유해 반응(20% 이상)은 메스꺼움, 피로, 기침, 설사, 두통이었습니다.
|
유해 반응 |
PROMACTA n = 43 (%) |
|
메스꺼움 |
33 |
|
피로 |
28 |
|
기침 |
23 |
|
설사 |
21 |
|
두통 |
21 |
|
사지 통증 |
19 |
|
발열 |
14 |
|
현기증 |
14 |
|
구강 인두 통증 |
14 |
|
복통 |
12 |
|
근육 경련 |
12 |
|
트랜스아미나제 증가 |
12 |
|
관절통 |
12 |
|
콧물 |
12 |
발진과 고빌리루빈혈증은 환자의 7%에서 보고되었으며, 백내장은 환자의 2%에서 보고되었습니다.
이 임상 시험에서, 총 빌리루빈이 1.5 x ULN보다 높은 상태에서 ALT 또는 AST가 3 x ULN보다 높은 경우는 환자의 5%에서 보고되었습니다. 총 빌리루빈이 1.5 x ULN보다 높은 경우는 환자의 14%에서 발생했습니다.
이 임상 시험에서, 환자들은 염색체 이상을 확인하기 위해 골수 천자를 받았습니다. 8명의 환자에서 치료 중 새로운 염색체 이상이 보고되었으며, 그 중 5명의 환자에서 7번 염색체에 복잡한 변화가 있었습니다.
6.2 시판 후 경험
PROMACTA의 시판 후 사용 중 다음과 같은 유해 반응이 확인되었습니다. 이러한 반응은 불확실한 규모의 모집단에서 자발적으로 보고되기 때문에, 빈도를 신뢰할 수 있게 추정하거나 약물 노출과의 인과 관계를 확립하는 것이 항상 가능한 것은 아닙니다.
피부 및 피하 조직 장애: 과색소침착 및 피부 황변을 포함한 피부 변색.
7 약물 상호작용
7.1 Polyvalent Cations (Chelation)
Eltrombopag은 음식, 미네랄 보충제 및 제산제에 함유된 다가 양이온(예: 철, 칼슘, 알루미늄, 마그네슘, 셀레늄, 아연)을 킬레이트합니다.
킬레이트로 인한 PROMACTA의 흡수 감소를 방지하려면 제산제, 유제품, 미네랄 보충제와 같이 다가 양이온이 포함된 약물이나 제품을 복용하기 최소 2시간 전이나 4시간 후에 PROMACTA를 복용하십시오. [복용량 및 투여(2.4), 임상 약리학(12.3) 참조].
7.2 Transporters
PROMACTA와 OATP1B1(예: 아토르바스타틴, 보센탄, 에제티미브, 플루바스타틴, 글리부리드, 올메사르탄, 피타바스타틴, 프라바스타틴, 로수바스타틴, 레파글리니드, 리팜피신, 심바스타틴산, SN-38[이리노테칸의 활성 대사체], 발사르탄) 또는 유방암 저항성 단백질(BCRP)(예: 이마티닙, 이리노테칸, 라파티닙, 메토트렉세이트, 미톡산트론, 로수바스타틴, 술파살라진, 토포테칸)의 기질인 약물을 병용 투여할 때는 주의하십시오. OATP1B1 또는 BCRP의 기질인 약물에 과도하게 노출되었는지 징후와 증상이 있는지 환자를 면밀히 모니터링하고, 적절한 경우 이러한 약물의 용량을 줄이는 것을 고려하십시오. PROMACTA를 사용한 임상 시험에서 로수바스타틴의 용량을 50% 줄이는 것이 권장되었습니다.
7.3 Protease Inhibitors
HIV Protease Inhibitors: PROMACTA를 로피나비르/리토나비르(LPV/RTV)와 병용 투여할 때 용량 조절은 권장되지 않습니다. 다른 HIV 프로테아제 억제제와의 약물 상호 작용은 평가되지 않았습니다.
Hepatitis C Virus Protease Inhibitors: PROMACTA를 보세프레비르 또는 텔라프레비르와 병용 투여할 때 용량 조절은 권장되지 않습니다. 다른 C형 간염 바이러스(HCV) 프로테아제 억제제와의 약물 상호 작용은 평가되지 않았습니다.
7.4 Peginterferon Alfa-2a/b Therapy
PROMACTA를 페그인터페론 알파-2a(PEGASYS®) 또는 -2b(PEGINTRON®)와 병용 투여할 때 용량 조절은 권장되지 않습니다.
8 특정 집단에서의 사용
8.1 임신
위험 요약
임산부에서 PROMACTA 사용에 대한 소수의 발표된 사례 보고 및 시판 후 경험에서 얻은 데이터는 주요 선천적 기형, 유산 또는 모체 또는 태아 결과에 대한 약물 관련 위험을 평가하기에 충분하지 않습니다. 동물 생식 및 발달 독성 연구에서 임신 랫트에 대한 엘트롬보파그의 경구 투여는 모체 독성 용량에서 배아 사망률과 태아 체중 감소를 초래했습니다. 이러한 효과는 75mg/일의 지속적 또는 만성 ITP 환자에서 곡선 아래 면적(AUC)을 기준으로 한 인간 임상 노출의 6배, 100mg/일의 만성 C형 간염 환자에서 AUC의 3배에 해당하는 노출을 초래하는 용량에서 관찰되었습니다. (데이터 참조).
지정된 모집단에 대한 주요 선천적 기형 및 유산의 추정 배경 위험은 알려져 있지 않습니다. 모든 임신에는 선천적 기형, 손실 또는 기타 부작용의 배경 위험이 있습니다. 미국 일반 인구에서 임상적으로 인식된 임신에서 주요 선천적 기형 및 유산의 추정 배경 위험은 각각 2%에서 4% 및 15%에서 20%입니다.
데이터
동물 데이터
초기 배아 발달 연구에서 암컷 랫트에 10, 20 또는 60mg/kg/일(각각 75mg/일의 ITP 환자에서 AUC를 기준으로 한 인간 임상 노출의 0.8, 2 및 6배, 100mg/일의 만성 C형 간염 환자에서 AUC를 기준으로 한 인간 임상 노출의 0.3, 1 및 3배)의 용량으로 경구 엘트롬보파그를 투여했습니다. 모체 독성을 유발한 최고 용량에서 착상 전 및 착상 후 손실 증가와 태아 체중 감소가 관찰되었습니다.
배아-태아 발달 연구에서 엘트롬보파그는 기관 형성 기간 동안 임신 랫트에 10, 20 또는 60mg/kg/일(각각 75mg/일의 ITP 환자에서 AUC를 기준으로 한 인간 임상 노출의 0.8, 2 및 6배, 100mg/일의 만성 C형 간염 환자에서 AUC를 기준으로 한 인간 임상 노출의 0.3, 1 및 3배)의 용량으로 경구 투여되었습니다. 모체 독성을 유발한 최고 용량에서 태아 체중 감소(6%에서 7%)와 경추 갈비뼈 존재의 약간 증가가 관찰되었습니다. 그러나 주요 구조적 기형의 증거는 관찰되지 않았습니다.
배아-태아 발달 연구에서 엘트롬보파그는 기관 형성 기간 동안 임신 토끼에 30, 80 또는 150mg/kg/일(각각 75mg/일의 ITP 환자에서 AUC를 기준으로 한 인간 임상 노출의 0.04, 0.3 및 0.5배, 100mg/일의 만성 C형 간염 환자에서 AUC를 기준으로 한 인간 임상 노출의 0.02, 0.1 및 0.3배)의 용량으로 경구 투여되었습니다. 태아 독성, 배아 사망률 또는 기형 발생의 증거는 관찰되지 않았습니다.
임신 랫트(F0)에서 태아 전 및 후 발달 독성 연구에서 경구 엘트롬보파그는 임신 6일부터 수유 20일까지 투여되었습니다. 20mg/kg/일(75mg/일의 ITP 환자에서 AUC를 기준으로 한 인간 임상 노출의 2배, 100mg/일의 만성 C형 간염 환자에서 AUC를 기준으로 한 인간 임상 노출과 유사)까지의 용량에서 모체 생식 기능 또는 자손(F1)의 발달에 대한 부작용은 관찰되지 않았습니다. 엘트롬보파그는 자손(F1)의 혈장에서 검출되었습니다. 새끼의 혈장 농도는 F0 어미에게 약물을 투여한 후 용량에 따라 증가했습니다.
8.2 수유
위험 요약
모유에서 엘트롬보파그 또는 그 대사산물의 존재, 모유 수유 아동에 대한 영향 또는 모유 생산에 대한 영향에 대한 데이터는 없습니다. 그러나 엘트롬보파그는 출산 후 10일에 수유 랫트의 새끼에서 검출되어 수유 중 전달 가능성을 시사합니다. PROMACTA로 인해 모유 수유 아동에게 심각한 부작용이 발생할 가능성이 있으므로 치료 중에는 모유 수유를 권장하지 않습니다.
8.3 생식 능력이 있는 여성 및 남성
피임
동물 생식 연구에 따르면 PROMACTA는 임산부에게 투여하면 태아에게 해를 끼칠 수 있습니다. 생식 능력이 있는 성적으로 활동적인 여성은 PROMACTA 치료 중 및 PROMACTA 치료 중단 후 최소 7일 동안 효과적인 피임법(임신율이 1% 미만인 방법)을 사용해야 합니다.
8.4 소아 사용
PROMACTA의 안전성 및 유효성은 지속적 또는 만성 ITP가 있는 1세 이상의 소아 환자와 IST 미경험 중증 재생 불량성 빈혈(h-ATG 및 사이클로스포린과 병용)이 있는 2세 이상의 소아 환자에서 확립되었습니다. ITP가 있는 1세 미만의 소아 환자에서의 안전성 및 유효성은 확립되지 않았습니다. 만성 C형 간염과 관련된 혈소판 감소증 및 난치성 중증 재생 불량성 빈혈이 있는 소아 환자에서의 안전성 및 유효성은 확립되지 않았습니다.
지속적 또는 만성 ITP가 있는 1세 이상의 소아 환자에서 PROMACTA의 안전성 및 유효성은 두 건의 이중맹검, 위약 대조 시험에서 평가되었습니다. [부작용(6.1), 임상 연구(14.1) 참조]. 엘트롬보파그의 약동학은 ITP가 있는 1세 이상의 소아 환자 168명에서 하루에 한 번 투여하여 평가되었습니다. [임상 약리학(12.3) 참조]. 1세 이상의 소아 환자에 대한 투약 권장 사항은 투약 및 투여(2.1)을 참조하십시오.
PROMACTA의 안전성 및 유효성은 2세 이상 소아 환자에서 중증 재생불량성 빈혈의 1차 치료를 위한 h-ATG 및 사이클로스포린과의 병용 투여에 대한 단일군, 공개 표지 시험에서 평가되었습니다. [유해 반응 (6.1), 임상 연구 (14.3) 참조]. 총 26명의 소아 환자(2세 ~ < 17세)가 평가되었으며, 12명의 어린이(2세 ~ < 12세)와 14명의 청소년(12세 ~ < 17세)이 포함되었습니다. 2세 이상 소아 환자에 대한 투약 권장 사항은 투약 및 투여 (2.3)을 참조하십시오. 중증 재생불량성 빈혈의 1차 치료를 위한 h-ATG 및 사이클로스포린과의 병용 투여에서 2세 미만 소아 환자에 대한 PROMACTA의 안전성 및 유효성은 아직 확립되지 않았습니다. 2세에서 16세 사이의 환자에서 69%의 환자가 심각한 유해 사건을 경험한 반면, 17세 이상의 환자에서는 42%였습니다. PROMACTA D1-M6 코호트에서 2세에서 11세 사이의 12명의 환자 중 6개월 평가에 도달하거나 조기에 중단한 환자의 경우, 6개월 시점의 완전 반응률은 8%였으며, 12세에서 16세 사이의 환자는 46%, 17세 이상의 환자는 50%였습니다.
8.5 노인
지속성 또는 만성 ITP에 대한 PROMACTA 50mg의 두 가지 무작위 대조 임상 시험에서 106명의 환자 중 22%가 65세 이상이었고, 9%가 75세 이상이었습니다. 만성 C형 간염 및 혈소판 감소증 환자에 대한 PROMACTA의 두 가지 무작위 대조 임상 시험에서 1439명의 환자 중 7%가 65세 이상이었고, 1% 미만이 75세 이상이었습니다. 중증 재생불량성 빈혈 치료를 위해 PROMACTA를 투여받은 196명의 환자 중 18%가 65세 이상이었고, 3%가 75세 이상이었습니다. 이러한 환자와 젊은 환자 사이에 안전성 또는 유효성에 대한 전반적인 차이는 관찰되지 않았습니다.
8.6 간 기능 장애
지속성 또는 만성 ITP 및 중증 재생불량성 빈혈 환자
간 기능 장애(Child-Pugh 등급 A, B, C)가 있는 지속성 또는 만성 ITP(성인 및 6세 이상 소아 환자만 해당) 또는 난치성 중증 재생불량성 빈혈 환자의 경우 PROMACTA의 초기 용량을 줄입니다. [투약 및 투여 (2.1, 2.3), 경고 및 주의 사항 (5.2), 임상 약리 (12.3) 참조].
이전에 확실한 면역 억제 요법을 받지 않은 중증 재생불량성 빈혈 환자를 대상으로 한 임상 시험에서 기준 ALT 또는 AST가 > 5 x ULN인 환자는 참여할 수 없었습니다. 간 기능 장애(Child-Pugh 등급 A, B, C)가 있는 환자가 중증 재생불량성 빈혈의 1차 치료를 위해 PROMACTA 치료를 시작하는 경우 초기 용량을 줄입니다. [투약 및 투여 (2.3), 경고 및 주의 사항 (5.2), 임상 약리 (12.3) 참조].
만성 C형 간염 환자
만성 C형 간염 및 간 기능 장애가 있는 환자의 경우 용량 조절은 권장되지 않습니다. [임상 약리 (12.3) 참조].
8.7 민족
ITP(성인 및 6세 이상 소아 환자만 해당) 또는 중증 재생불량성 빈혈이 있는 동아시아/동남아시아계 환자의 경우 PROMACTA의 초기 용량을 줄입니다. [투약 및 투여 (2.1, 2.3), 임상 약리 (12.3) 참조]. 만성 C형 간염이 있는 동아시아/동남아시아계 환자의 경우 PROMACTA의 초기 용량을 줄이는 것은 권장되지 않습니다. [임상 약리 (12.3) 참조].
10 과다 복용
과량 복용 시 혈소판 수치가 과도하게 증가하여 혈전증/색전증 합병증이 발생할 수 있습니다.
한 보고서에서 PROMACTA 5000mg을 섭취한 환자의 경우 섭취 후 13일째 혈소판 수치가 최대 929 x 109/L까지 증가했습니다. 환자는 또한 발진, 서맥, ALT/AST 상승 및 피로를 경험했습니다. 환자는 위세척, 경구 락툴로스, 정맥 수액, 오메프라졸, 아트로핀, 푸로세미드, 칼슘, 덱사메타손 및 혈장 교환술로 치료를 받았지만 비정상적인 혈소판 수치와 간 기능 검사 이상은 3주 동안 지속되었습니다. 2개월 추적 관찰 후 모든 사건은 후유증 없이 해결되었습니다.
과량 복용 시 칼슘, 알루미늄 또는 마그네슘 제제와 같은 금속 양이온 함유 제제를 경구 투여하여 엘트롬보파그를 킬레이트화하여 흡수를 제한하는 것을 고려하십시오. 혈소판 수치를 면밀히 모니터링하십시오. 투약 및 투여 권장 사항에 따라 PROMACTA 치료를 재개하십시오 [용법 및 용량 (2.1, 2.2) 참조].
11 설명
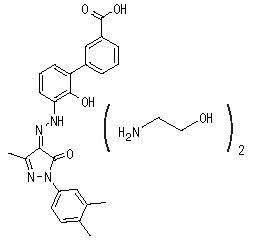
PROMACTA (엘트롬보파그) 정제는 경구 투여용 소분자 트롬보포이에틴(TPO) 수용체 작용제인 엘트롬보파그 올라민을 함유하고 있습니다.
엘트롬보파그 올라민은 비페닐 히드라존입니다. 엘트롬보파그 올라민의 화학명은 3′-{(2Z)-2-[1-(3,4-디메틸페닐)-3-메틸-5-옥소-1,5-디히드로-4H-피라졸-4-일리덴]히드라지노}-2′-하이드록시-3-비페닐카르복실산 – 2-아미노에탄올(1:2)입니다. 분자식은 C25H22N4O4 • 2(C2H7NO)이며, 엘트롬보파그 올라민의 분자량은 564.65 g/mol이고 엘트롬보파그 유리산의 분자량은 442.5 g/mol입니다. 엘트롬보파그 올라민의 구조식은 다음과 같습니다.
엘트롬보파그 올라민은 pH 1~7.4 범위의 수성 완충액에서 실질적으로 불용성이며 물에 약간 용해됩니다.
PROMACTA (엘트롬보파그) 정제는 엘트롬보파그 유리산과 동일한 양의 엘트롬보파그 올라민을 함유하고 있으며, 엘트롬보파그 유리산 12.5 mg, 25 mg, 50 mg 또는 75 mg에 해당합니다. PROMACTA 정제의 비활성 성분은 다음과 같습니다.
정제 코어: 마그네슘 스테아레이트, 만니톨, 미결정 셀룰로오스, 포비돈, 옥수수 전분.
코팅: FD&C 블루 No. 2 알루미늄 레이크(50mg 정제), FD&C 옐로우 No. 6 알루미늄 레이크(25mg 정제), 하이프로멜로오스, 산화철 블랙 및 산화철 레드(75mg 정제), 폴리에틸렌 글리콜 400, 폴리소르베이트 80(12.5mg 정제) 또는 이산화티타늄.
PROMACTA (엘트롬보파그) 경구 현탁액 포는 물에 재구성하면 적갈색 현탁액을 생성하는 적갈색에서 노란색 분말을 함유하고 있습니다. 각 포는 엘트롬보파그 유리산 12.5 mg 또는 25 mg에 해당하는 엘트롬보파그 올라민을 제공합니다. PROMACTA 경구 현탁액의 비활성 성분은 만니톨, 수크랄로스 및 잔탄 검입니다.
12 임상약리학
12.1 작용 기전
엘트롬보파그는 TPO 수용체 작용제로서 인간 TPO 수용체(cMpl로도 알려짐)의 막횡단 영역과 상호 작용하여 거핵구의 증식 및 분화를 유도하는 신호 전달 캐스케이드를 시작하여 혈소판 생성을 증가시킵니다.
12.2 약력학
임상 시험에서 PROMACTA로 치료한 결과, 반복 투여(매일) 후 혈소판 수치가 용량 의존적으로 증가했습니다. 혈소판 수치 증가는 투여 시작 후 약 2주 만에 최대에 도달했으며, PROMACTA 마지막 투여 후 약 2주 이내에 기준선으로 돌아왔습니다.
심장 전기 생리학
5일 동안 최대 150mg(최대 권장 용량)까지 투여한 경우, PROMACTA는 QT/QTc 간격을 유의미하게 연장시키지 않았습니다.
12.3 약동학
건강한 성인 피험자에서 엘트롬보파그는 50~150mg/일 용량 범위에서 용량 비례적으로 노출 증가를 보였습니다. 엘트롬보파그 AUC는 지속적 또는 만성 ITP 환자에서 건강한 피험자에 비해 약 1.7배 높았고, HCV 환자에서 약 2.8배 높았습니다. 1일 1회 투여 후 약 1주일 만에 정상 상태에 도달했으며, 75mg/일에서 기하 평균 축적 비율은 1.56(90% 신뢰 구간 1.20, 1.63)이었습니다. 엘트롬보파그 AUC는 면역 억제 요법을 받지 않은 확실한 중증 재생 불량성 빈혈 환자에서 건강한 피험자에 비해 약 3.2배 높았으며, 이는 ITP 환자 또는 건강한 피험자에 비해 상대적으로 노출이 높고 만성 C형 간염 환자와 유사한 노출을 나타냅니다. 경구 현탁액 형태의 엘트롬보파그는 정제 형태에 비해 AUC0-INF가 22% 높았습니다.
흡수
엘트롬보파그는 경구 투여 후 2~6시간 만에 최고 농도에 도달합니다. 75mg 용액 단일 용량 투여 후 약물 관련 물질의 경구 흡수는 최소 52%로 추정되었습니다.
음식의 영향
표준 고지방 아침 식사(876 칼로리, 지방 52g, 탄수화물 71g, 단백질 34g, 칼슘 427mg)는 엘트롬보파그 AUC0-INF를 약 59%, Cmax를 65% 유의미하게 감소시켰고, Tmax를 1시간 지연시켰습니다. 노출 감소는 주로 높은 칼슘 함량 때문입니다.
칼슘 함량이 낮은 식사(≤ 50mg 칼슘)는 칼로리 및 지방 함량에 관계없이 엘트롬보파그 혈장 노출에 유의미한 영향을 미치지 않았습니다.
건강한 성인 피험자에서 고칼슘, 중간 지방, 중간 칼로리 식사와 함께 엘트롬보파그 경구 현탁액 25mg 단일 용량을 투여했을 때 AUC0-INF 및 Cmax에 미치는 영향은 표 14에 제시되어 있습니다.
| a372 칼로리, 지방 9g, 칼슘 448mg. | ||
| 엘트롬보파그 경구 현탁액 용량 투여 시기 | 엘트롬보파그 혈장 AUC0-INF 감소 평균(90% CI) | 엘트롬보파그 혈장 Cmax 감소 평균(90% CI) |
| 고칼슘, 중간 지방, 중간 칼로리 식사와 함께 | 75%(71%, 88%) | 79%(76%, 82%) |
| 고칼슘, 중간 지방, 중간 칼로리 식사 후 2시간 | 47%(40%, 53%) | 48%(40%, 54%) |
| 고칼슘, 중간 지방, 중간 칼로리 식사 2시간 전 | 20%(9%, 29%) | 14%(2%, 25%) |
분포
방사성 동위원소 연구에 따르면 혈액 세포 내 엘트롬보파그 농도는 혈장 농도의 약 50%에서 79%입니다. 시험관 내 연구에 따르면 엘트롬보파그는 인간 혈장 단백질에 매우 높게 결합합니다(99% 이상). 엘트롬보파그는 BCRP의 기질이지만 P-당단백질(P-gp) 또는 OATP1B1의 기질은 아닙니다.
배설
건강한 피험자의 경우 엘트롬보파그의 혈장 제거 반감기는 약 21~32시간이고, ITP 환자의 경우 26~35시간입니다.
대사: 흡수된 엘트롬보파그는 광범위하게 대사되며, 주로 분해, 산화 및 글루쿠론산, 글루타티온 또는 시스테인과의 결합을 포함한 경로를 통해 이루어집니다. 시험관 내 연구에 따르면 CYP1A2와 CYP2C8은 엘트롬보파그의 산화적 대사를 담당합니다. UGT1A1과 UGT1A3은 엘트롬보파그의 글루쿠론화를 담당합니다.
배설: 엘트롬보파그의 주요 배설 경로는 대변(59%)이며, 투여량의 31%가 소변에서 발견됩니다. 대변에서 변하지 않은 엘트롬보파그는 투여량의 약 20%를 차지하며, 변하지 않은 엘트롬보파그는 소변에서 검출되지 않습니다.
특정 인구 집단
민족
ITP 또는 만성 C형 간염이 있는 동아시아/동남아시아계 환자의 엘트롬보파그 농도는 비아시아 피험자에 비해 50%에서 55% 더 높았습니다. [용법 및 용량(2.1, 2.3) 참조].
한 임상 약리학 시험에서 건강한 아프리카계 미국인 피험자의 엘트롬보파그 노출은 백인 피험자에서 관찰된 것보다 약 40% 더 높았고, 다른 세 가지 임상 약리학 시험에서는 유사했습니다. 아프리카계 미국인 민족이 엘트롬보파그 노출 및 관련 안전성 및 유효성에 미치는 영향은 확립되지 않았습니다.
간 기능 장애
PROMACTA(50mg)를 단일 투여한 후, 경증 간 기능 장애(Child-Pugh 등급 A) 환자의 혈장 엘트롬보파그 AUC0-INF는 정상 간 기능을 가진 피험자에 비해 41% 더 높았습니다. 혈장 엘트롬보파그 AUC0-INF는 중등도(Child-Pugh 등급 B) 및 중증 간 기능 장애(Child-Pugh 등급 C) 환자의 경우 정상 간 기능을 가진 피험자에 비해 약 2배 더 높았습니다. 엘트롬보파그의 반감기는 이러한 환자에서 2배 연장되었습니다. 이 임상 시험에서는 단백질 결합 효과를 평가하지 않았습니다.
만성 간 질환
혈소판 감소증 및 만성 간 질환이 있는 환자에게 엘트롬보파그를 반복 투여한 후, 경증 간 기능 장애는 혈장 엘트롬보파그 AUC(0-τ)가 87%에서 110% 더 높게 나타났고, 중등도 간 기능 장애는 혈장 엘트롬보파그 AUC(0-τ)가 약 141%에서 240% 더 높게 나타났습니다. 정상 간 기능을 가진 환자와 비교했습니다. 엘트롬보파그의 반감기는 경증 간 기능 장애 환자의 경우 3배, 중등도 간 기능 장애 환자의 경우 4배 연장되었습니다. 이 임상 시험에서는 단백질 결합 효과를 평가하지 않았습니다.
만성 C형 간염
PROMACTA로 치료받은 만성 C형 간염 환자는 건강한 피험자에 비해 혈장 AUC(0-τ) 값이 더 높았고, AUC(0-τ)는 Child-Pugh 점수가 높아짐에 따라 증가했습니다. 만성 C형 간염 및 경증 간 기능 장애가 있는 환자는 건강한 피험자에 비해 혈장 AUC(0-τ)가 약 100%에서 144% 더 높았습니다. 이 임상 시험에서는 단백질 결합 효과를 평가하지 않았습니다.
신장 기능 장애
PROMACTA(50mg)를 단일 투여한 후, 경증(Cockcroft-Gault 방정식으로 추정한 크레아티닌 청소율(CLCr): 50~80mL/분), 중등도(CLCr 30~49mL/분) 신장 기능 장애가 있는 피험자의 평균 총 혈장 엘트롬보파그 AUC0-INF는 건강한 피험자에 비해 32%에서 36% 낮았고, 중증(CLCr 30mL/분 미만) 신장 기능 장애가 있는 피험자의 경우 60% 낮았습니다. 신장 기능 장애가 결합되지 않은(활성) 엘트롬보파그 노출에 미치는 영향은 평가되지 않았습니다.
소아 환자
엘트롬보파그의 약동학은 두 가지 시험에서 ITP가 있는 1세 이상의 소아 환자 168명을 대상으로 하루에 한 번 투여하여 평가했습니다. 경구 투여 후 혈장 엘트롬보파그 명백한 청소율(CL/F)은 체중이 증가함에 따라 증가했습니다. ITP가 있는 동아시아/동남아시아계 소아 환자는 비아시아 환자에 비해 혈장 엘트롬보파그 AUC(0-τ) 값이 약 43% 더 높았습니다.
12~17세 소아 환자의 혈장 엘트롬보파그 AUC(0-τ) 및 Cmax는 성인에서 관찰된 것과 유사했습니다. ITP가 있는 소아 환자의 엘트롬보파그 약동학적 매개변수는 표 15에 나와 있습니다.
| aPK 매개변수는 기하 평균(95% CI)으로 표시됩니다. b인구 PK 사후 분석 추정치를 기반으로 합니다. |
||
|
Cmaxb |
AUC(0-τ)b |
|
|
연령 |
(mcg/mL) |
(mcg·hr/mL) |
|
성인(n = 108) |
7.03 |
101 |
|
(6.44, 7.68) |
(91.4, 113) |
|
|
12 to 17 years (n = 62) |
6.80 |
103 |
|
(6.17, 7.50) |
(91.1, 116) |
|
|
6 to 11 years (n = 68) |
10.3 |
153 |
|
(9.42, 11.2) |
(137, 170) |
|
|
1 to 5 years (n = 38) |
11.6 |
162 |
|
(10.4, 12.9) |
(139, 187) |
|
약물 상호 작용 연구
임상 연구
약물의 엘트롬보파그에 대한 영향
다가 양이온 함유 제산제의 엘트롬보파그에 대한 영향:
PROMACTA(75 mg) 단회 투여와 다가 양이온 함유 제산제(수산화알루미늄 1,524 mg, 탄산마그네슘 1,425 mg, 알긴산나트륨)의 병용 투여는 엘트롬보파그의 혈장 AUC0-INF 및 Cmax를 약 70% 감소시켰습니다. 알긴산나트륨의 이러한 상호 작용에 대한 기여는 알려져 있지 않습니다.
HIV 프로테아제 억제제의 엘트롬보파그에 대한 영향:
로피나비르 400 mg/리토나비르 100 mg(1일 2회) 반복 투여와 PROMACTA(100 mg) 단회 투여의 병용 투여는 엘트롬보파그의 혈장 AUC0-INF를 17% 감소시켰습니다.
HCV 프로테아제 억제제의 엘트롬보파그에 대한 영향:
임상 시험에서 건강한 성인 피험자에게 텔라프레비르(8시간마다 750 mg) 또는 보세프레비르(8시간마다 800 mg)를 반복 투여하고 PROMACTA(200 mg)를 단회 투여한 결과 엘트롬보파그의 혈장 AUC0-INF 또는 Cmax가 유의미하게 변하지 않았습니다.
시클로스포린의 엘트롬보파그에 대한 영향:
PROMACTA(50 mg) 단회 투여와 OATP 및 BCRP 억제제인 시클로스포린(200 mg 또는 600 mg) 단회 투여의 병용 투여는 엘트롬보파그의 혈장 AUC0-INF를 18%에서 24% 감소시키고 Cmax를 25%에서 39% 감소시켰습니다.
페길화 인터페론 알파-2a + 리바비린 및 페길화 인터페론 알파-2b + 리바비린의 엘트롬보파그에 대한 영향:
페길화 인터페론 알파 + 리바비린 요법의 존재는 엘트롬보파그의 청소율에 유의미한 영향을 미치지 않았습니다.
엘트롬보파그의 다른 약물에 대한 영향
엘트롬보파그의 시토크롬 P450 효소 기질에 대한 영향:
PROMACTA(1일 1회 75 mg, 7일간)를 다회 투여한 결과 인간에서 CYP1A2(카페인), CYP2C19(오메프라졸), CYP2C9(플루르비프로펜) 또는 CYP3A4(미다졸람)에 대한 프로브 기질 조합의 대사 억제 또는 유도가 발생하지 않았습니다.
엘트롬보파그의 로수바스타틴에 대한 영향:
PROMACTA(1일 1회 75 mg, 5일간)를 다회 투여하고 로수바스타틴(OATP1B1 및 BCRP 기질; 10 mg)을 단회 투여한 결과 로수바스타틴의 혈장 AUC0-INF가 55% 증가하고 Cmax가 103% 증가했습니다.
엘트롬보파그의 HCV 프로테아제 억제제에 대한 영향:
임상 시험에서 건강한 성인 피험자에게 텔라프레비르(8시간마다 750 mg) 또는 보세프레비르(8시간마다 800 mg)를 반복 투여하고 PROMACTA(200 mg)를 단회 투여한 결과 텔라프레비르 또는 보세프레비르의 혈장 AUC0-INF 또는 Cmax가 유의미하게 변하지 않았습니다.
시험관 내 연구
엘트롬보파그의 대사 효소에 대한 영향
엘트롬보파그는 CYP2C8, CYP2C9, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7 및 UGT2B15를 억제할 가능성이 있음을 보여주었습니다.
엘트롬보파그의 수송체에 대한 영향
엘트롬보파그는 OATP1B1 및 BCRP를 억제할 가능성이 있음을 보여주었습니다.
13 비임상 독성학
13.1 발암성, 돌연변이 유발성, 생식능력 저해
엘트롬보파그는 독특한 TPO 수용체 특이성으로 인해 랫드, 마우스 또는 개에서 혈소판 생성을 자극하지 않습니다. 이러한 동물에서 얻은 데이터는 인간의 효과를 완전히 모델링하지 않습니다.
엘트롬보파그는 마우스에서 최대 75mg/kg/일 또는 랫드에서 최대 40mg/kg/일(ITP 환자에서 75mg/일의 AUC 기준으로 인간 임상 노출의 최대 4배, 만성 C형 간염 환자에서 100mg/일의 AUC 기준으로 인간 임상 노출의 2배)의 용량에서 발암성이 없었습니다.
엘트롬보파그는 박테리아 돌연변이 분석 또는 랫드에서 두 가지 in vivo 분석(마이크로뉴클레어스 및 예정되지 않은 DNA 합성, ITP 환자에서 75mg/일의 Cmax 기준으로 인간 임상 노출의 10배, 만성 C형 간염 환자에서 100mg/일의 Cmax 기준으로 인간 임상 노출의 7배)에서 돌연변이 유발성 또는 염색체 이상 유발성이 없었습니다. in vitro 마우스 림프종 분석에서 엘트롬보파그는 약간 양성(돌연변이 빈도가 3배 미만 증가)이었습니다.
엘트롬보파그는 랫드에서 최대 20mg/kg/일(ITP 환자에서 75mg/일의 AUC 기준으로 인간 임상 노출의 2배, 만성 C형 간염 환자에서 100mg/일의 AUC 기준으로 인간 임상 노출과 유사)의 용량에서 암컷 생식능력에 영향을 미치지 않았습니다. 엘트롬보파그는 랫드에서 최대 40mg/kg/일(ITP 환자에서 75mg/일의 AUC 기준으로 인간 임상 노출의 3배, 만성 C형 간염 환자에서 100mg/일의 AUC 기준으로 인간 임상 노출의 2배)의 용량에서 수컷 생식능력에 영향을 미치지 않았습니다.
13.2 동물 약리학 및/또는 독성학
용량 및 시간 의존적인 방식으로 설치류에서 치료 관련 백내장이 발견되었습니다. ITP 환자에서 75mg/일의 AUC 기준으로 인간 임상 노출의 6배 이상, 만성 C형 간염 환자에서 100mg/일의 AUC 기준으로 인간 임상 노출의 3배 이상에서 마우스는 6주 후, 랫드는 28주 후에 백내장이 관찰되었습니다. ITP 환자에서 75mg/일의 AUC 기준으로 인간 임상 노출의 4배 이상, 만성 C형 간염 환자에서 100mg/일의 AUC 기준으로 인간 임상 노출의 2배 이상에서 마우스는 13주 후, 랫드는 39주 후에 백내장이 관찰되었습니다. [경고 및 주의 사항 (5.5) 참조].
마우스와 랫드에서 최대 14일 동안의 연구에서 일반적으로 사망률 및 질병과 관련된 노출에서 신장 세뇨관 독성이 관찰되었습니다. 신장 세뇨관 독성은 마우스에서 2년 동안의 경구 발암성 연구에서 25, 75 및 150mg/kg/일의 용량에서도 관찰되었습니다. 가장 낮은 용량에서의 노출은 ITP 환자에서 75mg/일의 AUC 기준으로 인간 임상 노출의 1.2배, 만성 C형 간염 환자에서 100mg/일의 AUC 기준으로 인간 임상 노출의 0.6배였습니다. 2년 연구에서 신장 변화와 관련된 노출보다 높은 노출에서 13주 후 마우스에서 유사한 효과가 관찰되지 않아 이러한 효과가 용량 및 시간 의존적임을 시사합니다.
14 임상 연구
14.1 지속적 또는 만성 ITP
성인: 지속적 또는 만성 ITP 성인 환자에서 PROMACTA의 효능 및 안전성은 3건의 무작위 배정, 이중맹검, 위약 대조 시험과 공개 라벨 연장 시험에서 평가되었습니다.
TRA100773B 연구와 TRA100773A 연구(각각 연구 773B 및 연구 773A로 지칭 [NCT00102739])에서 이전에 ITP 치료를 1회 이상 완료했고 혈소판 수가 30 x 109/L 미만인 환자는 최대 6주 동안 매일 PROMACTA 또는 위약을 투여받도록 무작위 배정되었으며, 그 후 6주 동안 치료를 중단했습니다. 시험 기간 동안 혈소판 수가 200 x 109/L를 초과하면 PROMACTA 또는 위약을 중단했습니다.
환자의 중간 연령은 50세였으며, 60%가 여성이었습니다. 환자의 약 70%는 이전에 ITP 치료를 2회 이상 받았으며(주로 코르티코스테로이드, 면역 글로불린, 리툭시맙, 세포 독성 치료, 다나졸 및 아자티오프린), 환자의 40%는 비장 절제술을 받았습니다. 모든 치료군에서 중간 기준 혈소판 수(약 18 x 109/L)는 유사했습니다.
연구 773B는 114명의 환자(2:1)를 PROMACTA 50mg 또는 위약군에 무작위 배정했습니다. 진단 이후 시간이 기록된 60명의 환자 중 약 17%는 진단 이후 시간이 3-12개월인 지속적 ITP 정의를 충족했습니다. 연구 773A는 117명의 환자(1:1:1:1)를 위약 또는 PROMACTA의 3가지 용량 요법 중 하나(30mg, 50mg 또는 75mg)에 무작위 배정했으며, 각각 매일 투여했습니다. 진단 이후 시간이 기록된 51명의 환자 중 약 14%는 지속적 ITP 정의를 충족했습니다.
이 시험에서 PROMACTA의 효능은 반응률로 평가되었으며, 반응률은 치료 기간 동안 언제든지 기준 혈소판 수가 30 x 109/L 미만에서 50 x 109/L 이상으로 변화하는 것으로 정의되었습니다(표 16).
| ap-값 < 0.001 for PROMACTA versus placebo. | ||
|
연구 |
PROMACTA 매일 50mg |
위약 |
|
773B |
43/73 (59%)a |
6/37 (16%) |
|
773A |
19/27 (70%)a |
3/27 (11%) |
PROMACTA에 대한 혈소판 수 반응은 비장 절제술을 받았거나 받지 않은 환자에서 유사했습니다. 일반적으로 PROMACTA 투여 시작 후 1주일 만에 혈소판 수 증가가 관찰되었으며, 최대 반응은 치료 2주 후에 관찰되었습니다. PROMACTA의 위약 및 50mg 용량군에서, 혈소판 수가 200 x 109/L를 초과하여 증가함에 따라 각각 3% 및 27%의 환자에서 시험 약물이 중단되었습니다. PROMACTA 50mg 용량으로 치료한 기간의 중앙값은 연구 773B에서 43일, 연구 773A에서 42일이었습니다.
지혈 과제를 받은 7명의 환자 중, 추가 ITP 약물은 위약군 환자 3명 중 3명에게 필요했고, PROMACTA로 치료받은 4명 중 0명에게 필요했습니다. 대부분의 지혈 과제는 수술 절차 때문이었습니다. 수혈이 필요한 출혈은 위약군 환자 1명에게 발생했고, PROMACTA로 치료받은 환자에게는 발생하지 않았습니다.
RAISE 연구(NCT00370331)에서, 197명의 환자가 무작위로 (2:1) PROMACTA 50mg을 1일 1회 (n = 135) 또는 위약 (n = 62)을 6개월 동안 투여받았으며, 이 기간 동안 개별 혈소판 수에 따라 PROMACTA 용량을 조정할 수 있었습니다. 진단 이후 시간이 기록된 145명의 환자 중 19%가 지속적인 ITP의 정의를 충족했습니다. 환자는 PROMACTA로 6주 동안 치료받은 후 동반 ITP 약물을 서서히 줄이거나 중단할 수 있었습니다. 환자는 임상적으로 필요한 경우 시험 기간 동안 언제든지 구제 치료를 받을 수 있었습니다.
PROMACTA와 위약으로 치료받은 환자의 중앙값 연령은 각각 47세와 52.5세였습니다. PROMACTA와 위약으로 치료받은 환자의 약 절반(각각 47%와 50%)은 무작위 배정 시 동반 ITP 약물(주로 코르티코스테로이드)을 투여받고 있었으며, 기준 혈소판 수가 15 x 109/L 이하였습니다(각각 50%와 48%). PROMACTA와 위약으로 치료받은 환자의 유사한 비율(각각 37%와 34%)이 이전에 비장 절제술을 받았습니다.
이 시험에서 PROMACTA의 효능은 위약에 비해 PROMACTA를 투여받은 환자의 혈소판 수가 50 x 109/L 이상 및 400 x 109/L 이하인 확률로 평가되었으며, 6개월 치료 기간 동안 환자 반응 프로파일에 기반했습니다. 26주 치료를 완료한 134명의 환자 중, 지속적인 혈소판 반응(26주 치료 기간의 마지막 8주 중 6주 동안 혈소판 수가 50 x 109/L 이상 및 400 x 109/L 이하이며, 언제든지 구제 약물을 투여받지 않은 경우)은 PROMACTA로 치료받은 환자의 60%에서 달성되었고, 위약으로 치료받은 환자의 10%에서 달성되었습니다(비장 절제술을 받은 환자: PROMACTA 51%, 위약 8%; 비장 절제술을 받지 않은 환자: PROMACTA 66%, 위약 11%). PROMACTA로 치료받은 환자 그룹의 반응자 비율은 모든 치료 중 방문에서 위약 치료 그룹의 7%와 19%에 비해 37%와 56% 사이였습니다. PROMACTA로 치료받은 환자는 6개월 치료 기간 동안 전체적으로 위약으로 치료받은 환자에 비해 혈소판 수가 50 x 109/L와 400 x 109/L 사이에 있을 가능성이 훨씬 높았습니다.
치료 결과는 시험에 등록된 모든 환자에 대해 표 17에 제시되어 있습니다.
|
결과 |
PROMACTA n = 135 |
위약 n = 62 |
|
혈소판 수 ≥ 50 x 109/L인 주의 평균 수 |
11.3 |
2.4 |
|
구제 치료가 필요한 경우, n (%) |
24 (18) |
25 (40) |
기저 시점에 다른 ITP 치료를 받고 있는 94명의 환자 중, PROMACTA를 투여받은 63명 중 37명(59%)과 위약군 31명 중 10명(32%)이 시험 기간 동안 동반 치료를 중단했습니다.
EXTEND 연구(NCT00351468)에서 PROMACTA를 사용한 이전 임상 시험을 완료한 환자들은 동반 ITP 약물의 용량을 줄이거나 필요성을 없애기 위한 시도가 이루어진 공개, 단일군 시험에 등록되었습니다. EXTEND에서 302명의 환자에게 PROMACTA가 투여되었으며, 218명의 환자가 1년을 완료했고, 180명의 환자가 2년을 완료했고, 107명의 환자가 3년을 완료했고, 75명의 환자가 4년을 완료했고, 34명의 환자가 5년을 완료했고, 18명의 환자가 6년의 치료를 완료했습니다. PROMACTA 투여 전 기저 혈소판 수의 중앙값은 19 x 109/L였습니다. 연구에서 1, 2, 3, 4, 5, 6, 7년의 중앙 혈소판 수는 각각 85 x 109/L, 85 x 109/L, 105 x 109/L, 64 x 109/L, 75 x 109/L, 119 x 109/L, 76 x 109/L였습니다.
소아 환자: 지속적 또는 만성 ITP가 있는 1세 이상의 소아 환자에서 PROMACTA의 효능과 안전성은 두 건의 이중맹검, 위약 대조 시험에서 평가되었습니다. 시험은 ITP 진단 이후 시간이 다릅니다. 최소 6개월 대 최소 12개월. 시험 기간 동안 용량은 2주마다 최대 1일 75mg까지 증가될 수 있습니다. 혈소판 수가 200 x 109/L를 초과하면 PROMACTA의 용량이 감소되었고, 400 x 109/L를 초과하면 중단 및 감소되었습니다.
PETIT2 연구(NCT01520909)에서 혈소판 수가 30 x 109/L 미만인(n = 92) 이전 ITP 치료에 내성이 있거나 재발한 환자들은 연령별로 계층화되어 PROMACTA(n = 63) 또는 위약(n = 29)에 무작위 배정되었습니다. 6세에서 17세 사이의 환자의 시작 용량은 27kg 이상인 경우 1일 1회 50mg, 27kg 미만인 경우 1일 1회 37.5mg으로 경구 정제로 투여되었습니다. 체중에 관계없이 6세에서 17세 사이의 동아시아/동남아시아 환자에게는 1일 1회 25mg의 감소된 용량이 사용되었습니다. 1세에서 5세 사이의 환자의 시작 용량은 1일 1회 1.2mg/kg(동아시아/동남아시아 환자의 경우 1일 1회 0.8mg/kg)으로 경구 현탁액으로 투여되었습니다.
13주 무작위 이중맹검 기간은 24주 공개 기간이 이어졌으며, 이 기간 동안 양쪽 군의 환자들은 PROMACTA를 투여받을 자격이 있었습니다.
환자의 중앙 연령은 9세였으며, 48%가 여성이었습니다. 환자의 약 62%가 기저 혈소판 수가 15 x 109/L 이하였으며, 이는 치료 군 간에 유사한 특징이었습니다. 이전 ITP 치료(주로 코르티코스테로이드 및 면역 글로불린)를 2회 이상 받은 환자의 비율은 PROMACTA를 투여받은 군에서 73%, 위약을 투여받은 군에서 90%였습니다. PROMACTA를 투여받은 군에서 4명의 환자가 비장 절제술을 받았습니다.
이 시험에서 PROMACTA의 효능은 무작위 이중맹검 기간 5주에서 12주 사이에 8주 중 6주 이상 혈소판 수가 ≥ 50 x 109/L(구제 치료 없이)인 대상자의 비율로 평가되었습니다(표 18).
| ap-값 = PROMACTA 대 위약 < 0.001. | ||
|
연령대 |
PROMACTA |
위약 |
|
전체 |
26/63 (41%)a |
1/29 (3%) |
|
12세에서 17세 |
10/24 (42%) |
1/10 (10%) |
|
6세에서 11세 |
11/25 (44%) |
0/13 (0%) |
|
1세에서 5세 |
5/14 (36%) |
0/6 (0%) |
PROMACTA로 치료받은 소아 환자의 경우 위약군(21%)에 비해 더 많은 환자(75%)가 구출 요법 없이 무작위 치료 첫 12주 동안 적어도 한 번 이상 혈소판 수가 50 x 109/L 이상이었습니다. PROMACTA로 치료받은 소아 환자는 무작위 이중맹검 기간 동안 위약군 환자(24% [7/29])에 비해 구출 요법이 필요한 경우가 적었습니다(19% [12/63]). 6주에서 8주(5주에서 12주 사이) 동안 구출 요법 없이 혈소판 반응(≥ 50 x 109/L)을 보인 환자의 경우 62%(16/26)가 PROMACTA 투여 시작 후 첫 2주 안에 초기 반응을 보였습니다.
환자는 시험의 공개 표지 단계에서만 기저 ITP 치료를 줄이거나 중단할 수 있었습니다. 기저 치료를 받고 있는 15명의 환자 중 53%(8/15)가 구출 요법 없이 주로 코르티코스테로이드를 포함한 동반 치료를 줄였습니다(n = 1) 또는 중단했습니다(n = 7).
PETIT 연구(NCT00908037)에서 혈소판 수가 30 x 109/L 미만인 이전 ITP 치료에 내성이 있거나 재발한 환자(n = 67)는 연령별로 계층화되어 PROMACTA(n = 45) 또는 위약(n = 22)에 무작위 배정되었습니다(2:1). 환자의 약 15%가 지속적인 ITP 정의를 충족했습니다. 12세에서 17세 환자의 시작 용량은 체중이나 인종에 관계없이 1일 1회 37.5mg이었습니다. 6세에서 11세 환자의 시작 용량은 27kg 이상인 경우 1일 1회 50mg, 27kg 미만인 경우 1일 1회 25mg으로 경구 정제로 투여되었습니다. 이 연령대의 동아시아/동남아시아 환자의 경우 1일 1회 25mg(27kg 이상)과 12.5mg(27kg 미만)의 감소된 용량이 사용되었습니다. 1세에서 5세 환자의 시작 용량은 1일 1회 1.5mg/kg(동아시아/동남아시아 환자의 경우 1일 1회 0.8mg/kg)으로 경구 현탁액으로 투여되었습니다.
7주 무작위 이중맹검 기간은 최대 24주까지 공개 표지 기간이 이어졌으며, 두 군의 환자 모두 PROMACTA를 투여받을 수 있었습니다.
환자의 중간 연령은 10세였으며, 60%가 여성이었습니다. 환자의 약 51%가 기저 혈소판 수가 15 x 109/L 이하였습니다. PROMACTA로 치료받은 군과 위약군 모두 이전 ITP 치료(주로 코르티코스테로이드와 면역 글로불린)를 2회 이상 받은 환자의 비율이 84%였습니다. PROMACTA로 치료받은 군에서 5명의 환자가 비장 절제술을 받았습니다.
이 시험에서 PROMACTA의 효능은 무작위 이중맹검 기간 1주에서 6주 사이에 적어도 한 번 이상 혈소판 수가 50 x 109/L 이상(구출 요법 없이)인 환자의 비율로 평가했습니다(표 19). PROMACTA에 대한 혈소판 반응은 연령대별로 일관되었습니다.
| ap-값 = PROMACTA 대 위약 0.011. | ||
|
연령대 |
PROMACTA |
위약 |
|
전체 |
28/45 (62%)a |
7/22 (32%) |
|
12세에서 17세 |
10/16 (62%) |
0/8 (0%) |
|
6세에서 11세 |
12/19 (63%) |
3/9 (33%) |
|
1세에서 5세 |
6/10 (60%) |
4/5 (80%) |
PROMACTA로 치료받은 소아 환자는 위약군 환자에 비해 무작위 배정 이중맹검 기간 동안 구제 치료가 필요한 경우가 적었습니다(13% [6/45] 대 50% [11/22]).
환자는 시험의 공개 표지 단계에서만 기본 ITP 치료를 줄이거나 중단할 수 있었습니다. 기본적으로 다른 ITP 치료를 받고 있는 13명의 환자 중 46%(6/13)는 주로 코르티코스테로이드인 동반 치료를 줄이거나(n = 3) 중단했으며(n = 3) 구제 치료가 필요하지 않았습니다.
14.2 만성 C형 간염 관련 혈소판 감소증
성인 만성 C형 간염 환자의 혈소판 감소증 치료에 대한 PROMACTA의 효능과 안전성은 두 건의 무작위 배정 이중맹검 위약 대조 시험에서 평가되었습니다. ENABLE1 연구(NCT00516321)는 항바이러스 치료에 페그인터페론 알파-2a(PEGASYS®)와 리바비린을 사용했고, ENABLE2 연구(NCT00529568)는 페그인터페론 알파-2b(PEGINTRON®)와 리바비린을 사용했습니다. 두 시험 모두 혈소판 수가 75 x 109/L 미만인 환자를 등록했으며, 혈소판 수, 선별 HCV RNA 및 HCV 유형에 따라 계층화했습니다. Child-Pugh 점수가 6점을 초과하는(B등급 및 C등급), 복수 병력 또는 간성 뇌병증이 있는 간 기능 저하 증거가 있는 환자는 제외되었습니다. 두 시험 모두 환자의 중간 연령은 52세였으며, 63%가 남성이었고, 74%가 백인이었습니다. 환자의 69%는 HCV 유형 1, 4, 6을 가졌고, 나머지는 유형 2와 3을 가졌습니다. 환자의 약 30%는 이전에 인터페론과 리바비린으로 치료를 받았습니다. 환자 대부분(90%)은 비침습적 검사에서 나타난 바와 같이 교량 섬유화 및 간경변증을 보였습니다. 두 치료군 모두 유사한 비율(95%)의 환자가 기본적으로 Child-Pugh A등급(점수 5~6)을 보였습니다. 두 치료군 모두 유사한 비율(2%)의 환자가 기본적으로 국제 표준화 비율(INR)이 1.7을 초과했습니다. 두 치료군 모두 중간 기본 혈소판 수(약 60 x 109/L)가 유사했습니다. 시험은 2단계로 구성되었습니다. 항바이러스 치료 전 단계와 항바이러스 치료 단계입니다. 항바이러스 치료 전 단계에서 환자는 ENABLE1의 경우 90 x 109/L 이상, ENABLE2의 경우 100 x 109/L 이상의 혈소판 수를 높이기 위해 공개 표지 PROMACTA를 투여받았습니다. PROMACTA는 처음 2주 동안 1일 1회 25mg으로 투여되었으며, 항바이러스 치료를 시작하기 위한 최적의 혈소판 수를 달성하기 위해 2~3주 간격으로 25mg씩 증량되었습니다. 환자가 공개 표지 PROMACTA를 투여받을 수 있는 최대 기간은 9주였습니다. 목표 혈소판 수를 달성하면 환자는 치료 전 단계가 끝날 때 동일한 용량의 PROMACTA 또는 위약을 무작위로(2:1) 투여받았습니다. PROMACTA는 각각의 처방 정보에 따라 페길화 인터페론과 리바비린과 함께 최대 48주 동안 투여되었습니다.
두 시험 모두 PROMACTA의 효능은 항바이러스 치료 종료 후 24주에 HCV-RNA가 검출되지 않은 환자의 비율로 정의되는 지속적인 바이러스 반응(SVR)으로 평가되었습니다. 목표 혈소판 수 90 x 109/L 이상을 달성하는 데 걸리는 중간 시간은 약 2주였습니다. 환자의 95%가 항바이러스 치료를 시작할 수 있었습니다.
두 시험 모두 PROMACTA로 치료받은 환자에서 SVR을 달성한 비율이 유의하게 높았습니다(표 20 참조). SVR을 달성한 환자 비율의 개선은 기본 혈소판 수(50 x 109/L 미만 대 50 x 109/L 이상)를 기반으로 한 하위 그룹에서 일관되었습니다. 기본 바이러스 부하가 높은(800,000 이상) 환자의 경우 SVR 비율은 PROMACTA의 경우 18%(82/452)였고, 위약의 경우 8%(20/239)였습니다.
| 약어: HCV, C형 간염 바이러스. aPROMACTA는 페그인터페론 알파-2a(유형 1/4/6의 경우 48주 동안 주 1회 180mcg; 유형 2 또는 3의 경우 24주 동안)와 리바비린(1일 2회 경구로 800~1,200mg)과 함께 투여되었습니다. bPROMACTA는 페그인터페론 알파-2b(유형 1/4/6의 경우 48주 동안 주 1회 1.5mcg/kg; 유형 2 또는 3의 경우 24주 동안)와 리바비린(1일 2회 경구로 800~1,400mg)과 함께 투여되었습니다. c목표 혈소판 수는 ENABLE1의 경우 ≥ 90 x 109/L, ENABLE2의 경우 ≥ 100 x 109/L였습니다. dp-값 < 0.05(PROMACTA 대 위약). |
||||
|
ENABLE1a |
ENABLE2b |
|||
|
항바이러스 치료 전 단계 |
n = 715 |
n = 805 |
||
|
목표 혈소판 수를 달성하고 항바이러스 치료를 시작한 환자의 비율c |
95% |
94% |
||
|
항바이러스 치료 단계 |
PROMACTA n = 450 % |
위약 n = 232 % |
PROMACTA n = 506 % |
위약 n = 253 % |
|
전반적인 SVRd |
23 |
14 |
19 |
13 |
|
HCV 유전형 2,3 |
35 |
24 |
34 |
25 |
|
HCV 유전형 1, 4, 6 |
18 |
10 |
13 |
7 |
PROMACTA로 치료받은 환자의 대부분(76%)은 위약군의 19%와 비교하여 50 x 109/L 이상의 혈소판 수를 유지했습니다. PROMACTA를 투여받은 환자는 위약군(45% 대 27%)과 비교하여 항바이러스 용량 감소가 필요하지 않은 비율이 더 높았습니다.
14.3 중증 재생불량성 빈혈
중증 재생불량성 빈혈의 1차 치료
h-ATG와 사이클로스포린과 병용한 PROMACTA는 이전에 ATG, 알렘투주맙 또는 고용량 사이클로포스파마이드로 면역억제 요법(IST)을 받지 않은 중증 재생불량성 빈혈 환자를 대상으로 단일군, 단일센터, 공개 라벨 순차적 코호트 시험(연구 ETB115AUS01T, 연구 US01T [NCT01623167]로 지칭)에서 조사되었습니다. 총 153명의 환자가 연구 US01T에서 3개의 순차적 코호트와 3번째 코호트의 연장에서 PROMACTA를 투여받았습니다. 여러 코호트는 동일한 PROMACTA 시작 용량을 투여받았지만 치료 시작일과 기간이 달랐습니다. 12세 이상 환자의 PROMACTA 시작 용량은 1일 1회 150mg(동아시아/동남아시아인의 경우 75mg 감량), 6~11세 소아 환자의 경우 1일 1회 75mg(동아시아/동남아시아인의 경우 37.5mg 감량), 2~5세 소아 환자의 경우 1일 1회 2.5mg/kg(동아시아/동남아시아인의 경우 1.25mg/kg 감량)이었습니다.
- 코호트 1(n = 30): 14일차부터 6개월까지(D14-M6) PROMACTA + h-ATG 및 사이클로스포린
- 코호트 2(n = 31): 14일차부터 3개월까지(D14-M3) PROMACTA + h-ATG 및 사이클로스포린
- 코호트 3 + 연장 코호트 [PROMACTA D1-M6 코호트](n = 92): 1일차부터 6개월까지(D1-M6) PROMACTA + h-ATG 및 사이클로스포린(6개월에 혈액학적 반응을 보인 모든 환자는 저용량 사이클로스포린(유지 용량)을 투여받을 자격이 있음)
PROMACTA 용량 감소는 혈소판 수 증가 및 간 기능 장애에 대해 시행되었습니다. 표 21에는 연구 US01T에서 PROMACTA와 병용하여 투여된 h-ATG와 사이클로스포린의 용량이 포함되어 있습니다.
코호트 3 + 연장 코호트의 데이터는 중증 재생불량성 빈혈 환자의 1차 치료에 대한 PROMACTA의 효능을 뒷받침합니다(표 22). 이 섹션에 제시된 결과는 코호트 3 및 연장 코호트(n = 92)의 결과를 나타냅니다.
| a사이클로스포린 용량은 위에 권장된 목표 골 농도를 달성하도록 조정되었습니다. 적절한 사이클로스포린 처방 정보를 참조하십시오. b이상적인 체중과 실제 체중의 중간값으로 계산되었습니다. |
|
|
약물 |
주요 시험에서 투여된 용량 |
|
말 항림프구 글로불린(h-ATG) |
6개월 치료 기간의 1일차부터 4일차까지 실제 체중을 기준으로 1일 40mg/kg을 정맥 주사로 투여 |
|
사이클로스포린a |
12세 이상 환자(총 일일 용량 6mg/kg/일) 1일차부터 6개월 동안 실제 체중을 기준으로 12시간마다 경구로 3mg/kg을 투여 체질량 지수가 35를 초과하는 20세 이상 환자 또는 체질량 지수가 95th 백분위수를 초과하는 12~20세 환자: 1일차부터 6개월 동안 조정된 체중b을 기준으로 12시간마다 경구로 3mg/kg을 투여 2~11세 환자(총 일일 용량 12mg/kg/일) 1일차부터 6개월 동안 실제 체중을 기준으로 12시간마다 경구로 6mg/kg을 투여 체질량 지수가 95th 백분위수를 초과하는 2~11세 환자: 1일차부터 6개월 동안 조정된 체중b을 기준으로 12시간마다 경구로 6mg/kg을 투여 |
|
사이클로스포린 |
6개월에 혈액학적 반응을 보인 환자 추가 18개월 동안 고정 용량으로 1일 2mg/kg을 경구로 투여 |
PROMACTA D1-M6 코호트에서 중앙값 나이는 28세(범위, 5~82세)였으며, 환자의 16%와 28%가 각각 ≥ 65세와 < 17세였습니다. 환자의 46%가 남성이었고, 대부분의 환자는 백인(62%)이었습니다. 체중이 12kg 이하이거나 ALT 또는 AST가 정상 상한치의 5배를 초과하는 환자는 시험에서 제외되었습니다.
h-ATG 및 사이클로스포린과 병용한 PROMACTA의 효능은 6개월 후 완전 혈액학적 반응을 기준으로 확립되었습니다. 완전 반응은 적어도 1주일 간격으로 2회 연속 혈액 검사에서 다음 3가지 값을 모두 충족하는 혈액학적 매개변수로 정의되었습니다. 절대 호중구 수(ANC) > 1000/mcL, 혈소판 수 > 100 x 109/L 및 헤모글로빈 > 10 g/dL. 부분 반응은 적어도 1주일 간격으로 2회 연속 혈액 검사에서 다음 2가지 값을 모두 충족하는 중증 재생불량성 빈혈의 표준 기준을 더 이상 충족하지 않는 혈액 수치로 정의되었습니다. ANC > 500/mcL, 혈소판 수 > 20 x 109/L 또는 적혈구 생성 수 > 60,000/mcL. 전체 반응률은 부분 반응 수와 완전 반응 수를 합한 것으로 정의됩니다.
| 약어: NE, 추정 불가. a6개월 평가에 도달하거나 조기에 철회된 환자 수는 백분율 계산의 분모입니다. b어느 시점에서든 반응자 수. |
|
|
PROMACTA D1-M6 + h-ATG + 사이클로스포린 |
|
|
6개월, na 전체 반응, n (%) [95% CI] 완전 반응, n (%) [95% CI] |
87 |
|
전체 반응의 중앙값 기간, nb |
70 |
|
개월 (95% CI) |
24.3 (21.4, NE) |
|
완전 반응의 중앙값 기간, nb |
46 |
|
개월 (95% CI) |
24.3 (23.0, NE) |
1년차(n = 78)의 전체 및 완전 혈액학적 반응률은 각각 56.4% 및 38.5%이고, 2년차(n = 62)는 각각 38.7% 및 30.6%입니다.
소아 환자
2세에서 16세 사이의 34명의 환자가 연구 US01T에 등록되었습니다. D1-M6 코호트에서 25명의 소아 환자 중 7명과 17명이 각각 6개월에 완전 반응과 전체 반응을 달성했습니다.
난치성 중증 재생불량성 빈혈
PROMACTA는 적어도 1개의 이전 면역억제 요법에 대한 반응이 불충분하고 혈소판 수가 30 x 109/L 이하인 중증 재생불량성 빈혈 환자 43명을 대상으로 단일군, 단일센터, 공개 표지 시험(연구 ETB115AUS28T, 연구 US28T [NCT00922883]로 지칭됨)에서 연구되었습니다. PROMACTA는 2주 동안 1일 1회 50mg의 초기 용량으로 투여되었으며, 2주 간격으로 1일 1회 최대 용량 150mg까지 증량되었습니다. 연구에서 PROMACTA의 효능은 치료 12주 후 평가된 혈액학적 반응으로 평가되었습니다. 혈액학적 반응은 다음 기준 중 1개 이상을 충족하는 것으로 정의되었습니다. 1) 혈소판 수가 기준선보다 20 x 109/L 증가하거나, 최소 8주 동안 수혈 독립성을 유지하는 안정적인 혈소판 수; 2) 헤모글로빈이 1.5g/dL 이상 증가하거나, 8주 연속으로 적혈구(RBC) 수혈이 4단위 이상 감소; 3) ANC가 100% 증가하거나, ANC가 0.5 x 109/L 이상 증가. 혈액학적 반응이 관찰되지 않으면 16주 후에 PROMACTA를 중단했습니다. 반응을 보인 환자는 시험의 연장 단계에서 치료를 계속했습니다.
치료받은 집단의 중간 연령은 45세(범위, 17세에서 77세)였으며, 56%가 남성이었습니다. 기준선에서 중간 혈소판 수는 20 x 109/L, 헤모글로빈은 8.4g/dL, ANC는 0.58 x 109/L, 절대 적혈구 생성 수는 24.3 x 109/L였습니다. 환자의 86%가 적혈구(RBC) 수혈 의존적이었고, 91%가 혈소판 수혈 의존적이었습니다. 대부분의 환자(84%)는 적어도 2개의 이전 면역억제 요법을 받았습니다. 기준선에서 3명의 환자가 염색체 이상을 보였습니다.
표 23은 효능 결과를 보여줍니다.
| a단일 및 다계통을 포함합니다. bNR = 사건 수가 적어 도달하지 못함(재발). |
|
|
결과 |
PROMACTA n = 43 |
|
반응률a, n (%) 95% CI (%) |
17 (40) (25, 56) |
|
반응 기간의 중간값(월)(95% CI) |
NRb (3.0, NRb) |
17명의 반응자에서 혈소판 수혈 없는 기간은 8일에서 1096일까지였으며 중간값은 200일이었고, RBC 수혈 없는 기간은 15일에서 1082일까지였으며 중간값은 208일이었습니다.
연장 단계에서 8명의 환자가 다계통 반응을 달성했습니다. 이 환자 중 4명은 이후 PROMACTA 치료를 점차 줄였고 반응을 유지했습니다(중간 추적 관찰: 8.1개월, 범위, 7.2개월에서 10.6개월).
16 제공/보관 및 취급 방법
16.1 정제
- 12.5mg 정제는 둥글고 양쪽으로 볼록하며, 흰색의 필름 코팅 정제로 한쪽 면에 “GS MZ1”과 12.5가 각인되어 있으며, 30정들이 병에 포장되어 있습니다: NDC 0078-0684-15
- 25mg 정제는 둥글고 양쪽으로 볼록하며, 주황색의 필름 코팅 정제로 한쪽 면에 “GS NX3”과 25가 각인되어 있으며, 30정들이 병에 포장되어 있습니다: NDC 0078-0685-15
- 50mg 정제는 둥글고 양쪽으로 볼록하며, 파란색의 필름 코팅 정제로 한쪽 면에 “GS UFU”와 50이 각인되어 있으며, 다음과 같이 포장되어 있습니다:
- 14정들이 병 NDC 0078-0686-55
- 30정들이 병 NDC 0078-0686-15
- 75mg 정제는 둥글고 양쪽으로 볼록하며, 분홍색의 필름 코팅 정제로 한쪽 면에 “GS FFS”와 75가 각인되어 있으며, 30정들이 병에 포장되어 있습니다: NDC 0078-0687-15
20°C~25°C(68°F~77°F)의 실온에 보관하십시오. 15°C~30°C(59°F~86°F)의 온도에서도 보관 가능합니다 [USP 제어 실온 참조]. 원래 병에 보관하십시오.
16.2 경구 현탁액용
- 경구 현탁액용 12.5mg은 붉은 갈색에서 노란색의 분말로 단일 용량 포장으로 되어 있으며, 40cc 재구성 용기, 주사기 포트 기능이 있는 나사식 마개, 30개의 일회용 경구 투약 주사기가 함께 포장된 키트에 들어 있습니다.
각 키트(NDC 0078-0972-61)에는 30개의 포장이 들어 있습니다: NDC 0078-0972-19
- 경구 현탁액용 25mg은 붉은 갈색에서 노란색의 분말로 단일 용량 포장으로 되어 있으며, 40cc 재구성 용기, 주사기 포트 기능이 있는 나사식 마개, 30개의 일회용 경구 투약 주사기가 함께 포장된 키트에 들어 있습니다.
각 키트(NDC 0078-0697-61)에는 30개의 포장이 들어 있습니다: NDC 0078-0697-19
20°C~25°C(68°F~77°F)의 실온에 보관하십시오. 15°C~30°C(59°F~86°F)의 온도에서도 보관 가능합니다 [USP 제어 실온 참조]. 재구성 후에는 즉시 투여해야 하며, 20°C~25°C(68°F~77°F)의 온도에서 최대 30분 동안 보관할 수 있습니다. 15°C~30°C(59°F~86°F)의 온도에서도 보관 가능합니다 [USP 제어 실온 참조]. 30분 이내에 사용하지 않으면 버리십시오.
17 환자 상담 정보
환자 또는 보호자에게 FDA 승인 환자 라벨(약물 안내 및 사용 지침)을 읽도록 알려주십시오.
치료 전에 환자는 PROMACTA에 대한 다음 위험 및 고려 사항을 완전히 이해하고 알고 있어야 합니다.
위험
간 독성
- PROMACTA 치료는 간담도계 실험실 이상과 관련이 있을 수 있습니다 [경고 및 주의 사항(5.2) 참조].
- 만성 C형 간염 및 간경변 환자에게 알파 인터페론 치료와 함께 PROMACTA를 투여할 경우 간 기능 저하 위험이 있을 수 있음을 알려주십시오 [경고 및 주의 사항(5.1) 참조].
- 환자에게 다음과 같은 간 문제 증상을 의료 서비스 제공자에게 즉시 보고해야 함을 알려주십시오 [경고 및 주의 사항(5.2) 참조].
- 피부 또는 눈의 흰자위가 노랗게 변색(황달)
- 소변 색깔이 비정상적으로 어두워짐
- 비정상적인 피로감
- 오른쪽 위 복부 통증
- 혼란
- 복부 팽만(복부)
PROMACTA 중단 시 출혈 위험
- 환자에게 PROMACTA를 중단하면 특히 환자가 항응고제 또는 항혈소판제를 복용하는 동안 PROMACTA를 중단하면 혈소판 감소증과 출혈 위험이 재발할 수 있음을 알려주십시오. 환자에게 PROMACTA 치료 중에는 출혈 위험을 증가시킬 수 있는 상황이나 약물을 피해야 함을 알려주십시오.
혈전증/색전증 합병증
- 환자에게 PROMACTA를 너무 많이 복용하면 혈소판 수치가 과도하게 증가하여 혈전증/색전증 합병증 위험이 발생할 수 있음을 알려주십시오 [경고 및 주의 사항(5.4) 참조].
백내장
- 환자에게 PROMACTA 투여 전에 기저 안과 검사를 받고 치료 중에 백내장 징후 및 증상을 모니터링하도록 알려주십시오 [경고 및 주의 사항(5.5) 참조].
약물 상호 작용
- 환자에게 PROMACTA를 철, 칼슘, 알루미늄, 마그네슘, 셀레늄 및 아연과 같은 다가 양이온을 함유한 칼슘이 풍부한 음식, 미네랄 보충제 및 제산제보다 최소 2시간 전 또는 4시간 후에 복용하도록 알려주십시오 [투여 및 관리(2.4), 약물 상호 작용(7.1) 참조].
수유
- 여성에게 PROMACTA 치료 중에는 모유 수유를 하지 말라고 알려주십시오 [특정 인구 집단에서의 사용(8.2) 참조].
PROMACTA 투여
- 지속적 또는 만성 ITP 환자의 경우 PROMACTA 치료는 출혈 위험을 줄이기 위해 필요에 따라 혈소판 수치를 50 x 109/L 이상으로 유지하도록 투여됩니다 [적응증 및 사용(1.1) 참조].
- 만성 C형 간염 환자의 경우 PROMACTA 치료는 페길화 인터페론 및 리바비린으로 항바이러스 치료를 시작하고 유지하기 위해 필요한 혈소판 수치를 달성하고 유지하도록 투여됩니다 [적응증 및 사용(1.2) 참조].
- 환자에게 PROMACTA를 식사 없이 또는 칼슘 함량이 낮은 식사(≤ 50 mg)와 함께, 다른 약물(예: 제산제) 및 칼슘이 풍부한 음식보다 최소 2시간 전 또는 4시간 후에 복용하도록 알려주십시오 [투여 및 관리(2.4) 참조].
- 경구 현탁액을 사용하기 전에 환자 또는 보호자가 적절한 투여량, 준비 및 투여에 대한 교육을 받았는지 확인하십시오 [투여 및 관리(2.4) 참조].
- 환자 또는 보호자에게 전체 용량을 얻기 위해 몇 개의 포를 투여해야 하는지 알려주십시오 [사용 지침 참조].
- 환자 또는 보호자에게 PROMACTA 경구 현탁액의 각 용량을 준비할 때 새 경구 투여 주사기를 사용하도록 알려주십시오 [사용 지침 참조].
다음은 각 소유주의 등록 상표입니다: PEGASYS/Hoffmann-La Roche Inc.; PEGINTRON/Schering Corporation.
판매원:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
© Novartis
T2023-13
약물 안내문
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: March 2023 |
| MEDICATION GUIDE | |
| PROMACTA® (pro-MAC-ta) (eltrombopag) tablets |
PROMACTA® (pro-MAC-ta) (eltrombopag) for oral suspension |
| PROMACTA에 대해 알아야 할 가장 중요한 정보는 무엇입니까?
PROMACTA는 다음을 포함한 심각한 부작용을 일으킬 수 있습니다. 간 문제:
다음과 같은 간 문제 증상이 나타나면 즉시 의료 서비스 제공자에게 알리십시오. |
|
|
|
| PROMACTA의 다른 부작용에 대해서는 “PROMACTA의 가능한 부작용은 무엇입니까?”를 참조하십시오. | |
| PROMACTA는 무엇입니까? PROMACTA는 지속적이거나 만성적인 면역 혈소판 감소증(ITP)으로 인해 혈소판 수치가 낮은 1세 이상의 성인 및 어린이를 치료하는 데 사용되는 처방약으로, ITP 치료를 위한 다른 약물이나 비장 절제술이 충분히 효과적이지 않은 경우에 사용됩니다. PROMACTA는 또한 다음과 같은 사람들을 치료하는 데 사용됩니다.
PROMACTA는 출혈 위험을 줄이기 위해 혈소판 수치를 높이기 위해 사용됩니다. PROMACTA는 혈소판 수치를 정상으로 만들기 위해 사용되지 않습니다. PROMACTA는 골수이형성 증후군(MDS)이라는 전암 상태가 있는 사람이나 특정 다른 의학적 상태 또는 질병으로 인해 혈소판 수치가 낮은 사람에게는 사용되지 않습니다. PROMACTA가 만성 C형 간염 치료를 위한 다른 항바이러스 약물과 함께 사용할 때 안전하고 효과적인지 여부는 알려져 있지 않습니다. PROMACTA가 다음과 같은 어린이에게 안전하고 효과적인지 여부는 알려져 있지 않습니다.
|
|
PROMACTA를 복용하기 전에 다음과 같은 경우를 포함하여 모든 의학적 상태에 대해 의료 서비스 제공자에게 알리십시오.
특히 다음과 같은 약물을 복용하는 경우 의료 서비스 제공자에게 알리십시오.
특정 약물은 PROMACTA가 제대로 작동하지 못하게 할 수 있습니다. 다음과 같은 제품을 복용하기 최소 2시간 전 또는 4시간 후에 PROMACTA를 복용하십시오.
복용하는 약물이 위에 나열된 약물인지 확실하지 않으면 의료 서비스 제공자에게 문의하십시오. |
|
PROMACTA는 어떻게 복용해야 하나요?
|
|
| PROMACTA를 복용하는 동안 무엇을 피해야 하나요?
출혈 위험을 높일 수 있는 상황과 약물을 피하십시오. |
|
| PROMACTA의 가능한 부작용은 무엇인가요?
PROMACTA는 다음을 포함한 심각한 부작용을 유발할 수 있습니다.
성인과 어린이에게서 PROMACTA의 가장 흔한 부작용은 다음과 같습니다. |
|
|
|
| 실험실 검사에서 골수 세포에 비정상적인 변화가 나타날 수 있습니다. 귀찮거나 사라지지 않는 부작용이 있으면 의료 서비스 제공자에게 알리십시오. 이것들은 PROMACTA의 모든 가능한 부작용이 아닙니다. 자세한 내용은 의료 서비스 제공자 또는 약사에게 문의하십시오. 부작용에 대한 의학적 조언은 의사에게 문의하십시오. 부작용을 FDA에 보고할 수 있습니다. 1-800-FDA-1088. |
|
| PROMACTA 정제와 PROMACTA 구강 현탁액은 어떻게 보관해야 하나요?
정제:
구강 현탁액:
PROMACTA와 모든 약물은 어린이의 손이 닿지 않는 곳에 보관하십시오. |
|
| PROMACTA의 안전하고 효과적인 사용에 대한 일반 정보
약물은 때때로 약물 안내서에 나열된 목적 이외의 목적으로 처방됩니다. PROMACTA는 처방받지 않은 질환에 사용하지 마십시오. PROMACTA를 다른 사람에게 주지 마십시오. 같은 증상이 있어도 해로울 수 있습니다. 의료 서비스 제공자 또는 약사에게 의료 전문가를 위해 작성된 PROMACTA에 대한 정보를 요청할 수 있습니다. |
|
| PROMACTA에 어떤 성분이 들어 있나요? 정제 활성 성분: 엘트롬보파그 올라민 비활성 성분:
경구 현탁액용 판매원: 노바티스 파마슈티컬스 코퍼레이션, 이스트 해노버, 뉴저지 07936 |
|
T2023-14
사용 지침
PROMACTA® [pro-MAC-ta]
(엘트롬보파그)
경구 현탁액용
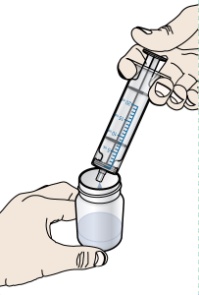
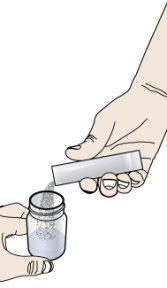
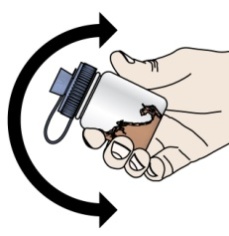
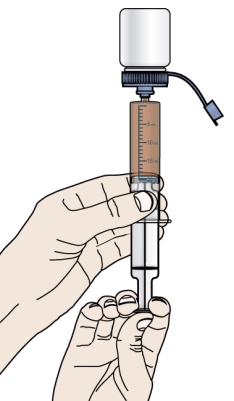
PROMACTA 경구 현탁액을 혼합하고 투여하는 방법에 대한 모든 사용 지침을 읽고 아래 단계를 따르십시오.
PROMACTA 경구 현탁액을 복용하기 전에 알아야 할 중요한 정보:
- PROMACTA 경구 현탁액을 복용하거나 다른 사람에게 투여하기 전에 PROMACTA 경구 현탁액을 올바르게 혼합하고 투여하는 방법을 보여주십시오. 의료 서비스 제공자 또는 간호사가 PROMACTA 경구 현탁액을 올바르게 혼합하고 투여하는 방법을 보여줄 것입니다.
- PROMACTA 경구 현탁액은 차갑거나 찬물에만 혼합해야 합니다. 경구 현탁액을 준비할 때 뜨거운 물을 사용하지 마십시오.
- 물과 혼합한 후 즉시 현탁액을 투여하십시오. 30분 이내에 투여하지 않으면 새로 혼합해야 합니다. 사용하지 않은 혼합물은 쓰레기통에 버리십시오. 배수구에 버리지 마십시오.
- PROMACTA 경구 현탁액이 피부에 닿으면 즉시 비누와 물로 씻으십시오. 피부 반응이 나타나거나 궁금한 사항이 있으면 의료 서비스 제공자에게 연락하십시오. 가루나 액체를 쏟았을 경우 12단계의 청소 지침을 따르십시오.
- 자녀에게 PROMACTA를 혼합하거나 투여하는 방법에 대한 질문이 있거나 키트의 공급품이 손상되거나 분실된 경우 의료 서비스 제공자 또는 약사에게 문의하십시오.
- 다시 사용하지 마십시오. PROMACTA 경구 현탁액을 준비할 때마다 새로 사용하는 일회용 경구 투약 주사기를 사용하십시오.
- 30개의 포장을 모두 사용한 후에는 남은 공급품(혼합 병, 뚜껑이 있는 뚜껑 및 경구 투약 주사기)을 모두 쓰레기통에 버리십시오.
각 PROMACTA 경구 현탁액 키트에는 다음과 같은 공급품이 들어 있습니다.
|
PROMACTA 경구 현탁액 30개 포장 |
 |
|
뚜껑과 뚜껑이 있는 재사용 가능한 혼합 병 1개 |
 |
|
일회용 20mL 경구 투약 주사기 30개 |
 |
경구 현탁액 PROMACTA 용량을 투여하려면 다음이 필요합니다.
키트에서:
- 처방된 수의 포장
- 뚜껑과 마개가 있는 재사용 가능한 혼합 병. 참고: 크기가 작기 때문에 마개가 어린 아이의 기도 폐쇄 위험을 초래할 수 있습니다.
- 일회용 20mL 경구 투약 주사기(PROMACTA 경구 현탁액 용량을 준비할 때마다 새(일회용) 경구 투약 주사기를 사용하십시오)
키트에 포함되지 않은 것:
- 깨끗한 유리잔 또는 컵에 담긴 식수
- 포장을 자르기 위한 가위
- 종이 타월 또는 일회용 천
- 일회용 장갑(선택 사항)
T2020-61
주요 디스플레이 패널
NDC 0078-0684-15
처방전에 의해서만 구입 가능
Promacta®
(eltrombopag) 정제
12.5 mg*
정제를 통째로 삼켜야 합니다.
정제를 쪼개거나 씹거나 부수지 마십시오.
약물 안내서를 함께 제공하거나 별도로 제공하십시오.
NOVARTIS
30 정

주요 디스플레이 패널
NDC 0078-0685-15
처방전에 의해서만 구입 가능
Promacta®
(eltrombopag) 정제
25 mg*
정제를 통째로 삼켜야 합니다.
정제를 쪼개거나, 씹거나, 부수지 마십시오.
약물 안내서를 함께 제공하거나 별도로 제공하십시오.
NOVARTIS
30 정

주요 디스플레이 패널
NDC 0078-0686-15
Rx only
Promacta®
(eltrombopag) Tablets
50 mg*
정제를 통째로 삼키십시오. 정제를 나누거나, 씹거나, 부수지 마십시오.
첨부된 또는 별도로 제공된 복약 안내서와 함께 제공하십시오.
NOVARTIS
30 Tablets

주요 디스플레이 패널
NDC 0078-0687-15
Rx only
Promacta®
(eltrombopag) Tablets
75 mg*
알정을 통째로 삼키십시오. 정제를 쪼개거나, 씹거나, 부수지 마십시오.
첨부되거나 별도로 제공된 복약 안내서와 함께 제공하십시오.
NOVARTIS
30 Tablets

주요 디스플레이 패널
NDC 0078-0972-61
처방전 의약품
Promacta®
(엘트롬보파그)
경구용 현탁액
12.5 mg
약물 안내서를 함께 제공하거나 별도로 제공하십시오.
30 포
NOVARTIS

주요 디스플레이 패널
NDC 0078-0697-61
처방전 의약품
Promacta®
(엘트롬보파그)
경구용 현탁액
25 mg
약물 안내서를 함께 제공하거나 별도로 제공하십시오.
30 포
NOVARTIS
