의약품 제조업체: Amneal Pharmaceuticals LLC (Updated: 2023-11-22)
처방 정보 요약
최초 미국 승인: 2001년
적응증 및 용법
Imatinib mesylate 정제는 다음의 치료를 위한 키나제 억제제입니다:
- 필라델피아 염색체 양성 만성 골수성 백혈병(Ph+ CML) 만성기의 새로 진단된 성인 및 소아 환자. (1.1)
- 필라델피아 염색체 양성 만성 골수성 백혈병(Ph+ CML) 급성기(BC), 가속기(AP) 또는 인터페론-알파 요법 실패 후 만성기(CP) 환자. (1.2)
- 재발 또는 불응성 필라델피아 염색체 양성 급성 림프구성 백혈병(Ph+ ALL) 성인 환자. (1.3)
- 화학요법과 병용하는 새로 진단된 필라델피아 염색체 양성 급성 림프구성 백혈병(Ph+ ALL) 소아 환자. (1.4)
- 혈소판 유래 성장 인자 수용체(PDGFR) 유전자 재배열과 관련된 골수이형성/골수증식성 질환(MDS/MPD) 성인 환자. (1.5)
- D816V c-Kit 변이가 없거나 c-Kit 변이 상태를 알 수 없는 공격적 전신 비만세포증(ASM) 성인 환자. (1.6)
- FIP1L1-PDGFRα 융합 키나제(변이 분석 또는 형광 제자리 하이브리드화[FISH] CHIC2 대립유전자 결실 입증)를 가진 하이퍼호산구증(HES) 및/또는 만성 호산구성 백혈병(CEL) 성인 환자, 그리고 FIP1L1-PDGFRα 융합 키나제 음성 또는 알 수 없는 HES 및/또는 CEL 환자. (1.7)
- 절제 불가능하고 재발성 및/또는 전이성 피부섬유육종 돌기(DFSP) 성인 환자. (1.8)
- Kit(CD117) 양성 절제 불가능 및/또는 전이성 악성 위장관 간질종(GIST) 환자. (1.9)
- Kit(CD117) 양성 GIST 절제 후 성인 환자의 보조 요법. (1.10)
투여 및 투여량
- Ph+ CML CP 성인 (2.2): 1일 400mg
- Ph+ CML AP 또는 BC 성인 (2.2): 1일 600mg
- Ph+ CML CP 소아 (2.3): 1일 340mg/m2
- Ph+ ALL 성인 (2.4): 1일 600mg
- Ph+ ALL 소아 (2.5): 1일 340mg/m2
- MDS/MPD 성인 (2.6): 1일 400mg
- ASM 성인 (2.7): 1일 100mg 또는 400mg
- HES/CEL 성인 (2.8): 1일 100mg 또는 400mg
- DFSP 성인 (2.9): 1일 800mg
- 전이성 및/또는 절제 불가능 GIST 성인 (2.10): 1일 400mg
- GIST 성인 보조 요법 (2.11): 1일 400mg
- 경증 내지 중등증 간장애 환자 (2.12): 1일 400mg
- 중증 간장애 환자 (2.12): 1일 300mg
Imatinib mesylate 정제의 모든 용량은 식사와 함께 물 한 큰 잔과 함께 복용해야 합니다. 400mg 또는 600mg 용량은 1일 1회 투여하고, 800mg 용량은 400mg를 1일 2회 투여합니다. Imatinib mesylate 정제를 물 또는 사과 주스에 녹여 삼키기 어려운 환자에게 투여할 수 있습니다. 1일 800mg 이상 용량은 철에 대한 노출을 줄이기 위해 400mg 정제를 사용해야 합니다.
제형 및 강도
정제(할선): 100mg 및 400mg (3)
금기 사항
없음. (4)
경고 및 주의사항
- 부종 및 심각한 체액 저류가 발생할 수 있습니다. 환자의 체중을 정기적으로 측정하고, 예기치 않은 급격한 체중 증가 시 약물 중단 및 이뇨제를 투여하십시오. (5.1, 6.1)
- 특히 빈혈, 중성구 감소증, 혈소판 감소증 등의 혈구 감소증이 발생할 수 있습니다. 용량 감량, 용량 중단 또는 투여 중단으로 관리하십시오. 첫 달에는 매주, 둘째 달에는 격주로, 그 후에는 주기적으로 전체 혈구 수를 검사하십시오. (5.2)
- 특히 동반 질환과 위험 요인이 있는 환자에서 심각한 울혈성 심부전 및 좌심실 기능 장애가 보고되었습니다. 심장 질환이나 심부전 위험인자가 있는 환자를 모니터링하고 치료하십시오. (5.3)
- 치명적일 수 있는 심각한 간독성이 발생할 수 있습니다. 치료 시작 전과 그 후 매달 또는 임상적으로 지시된 대로 간기능을 평가하십시오. 간기능 장애와 관련이 있는 것으로 알려진 화학요법제와 병용 시 간기능을 모니터링하십시오. (5.4)
- 신규 CML 및 GIST(위장관기질종양) 환자의 임상시험에서 3/4등급 출혈이 보고되었습니다. GIST의 경우 위장관 종양 부위가 출혈의 원인일 수 있습니다. (5.5)
- 일부 치명적인 위장관 천공이 보고되었습니다. (5.6)
- 고호산구증후군, 골수증식성질환/골수증식성질환, 혈관신경성부종 등 높은 호산구 수치와 관련된 질환에서 이 약 투여 시작 시 심장성 쇽/좌심실 기능 장애가 발생할 수 있습니다. (5.7)
- 다형홍반, 스티븐스-존슨 증후군 등의 수포성 피부 반응이 이 약 사용 시 보고되었습니다. (5.8)
- 갑상선 절제술을 받은 환자에서 레보치록신 대체 요법 중 갑상선기능저하증이 보고되었습니다. 이러한 환자에서 TSH 수치를 주의 깊게 모니터링하십시오. (5.9)
- 임신 중 이 약을 복용하면 태아에 해를 끼칠 수 있습니다. 환자들에게 태아에 대한 잠재적 위험을 알리고 효과적인 피임법을 사용하도록 하십시오. (5.10, 8.1)
- 이 약을 복용하는 아동 및 청소년에서 성장 지연이 보고되었습니다. 이 약 치료 중인 아동의 성장을 주의 깊게 모니터링하는 것이 권장됩니다. (5.11, 6.2)
- 종양 용해 증후군에 주의하십시오. 주의 깊게 모니터링하는 것이 권장됩니다. (5.12)
- 이 약 복용 시 운전 및 기계 조작 사고가 보고되었습니다. 환자에게 주의를 주십시오. (5.13)
- 신독성이 발생할 수 있습니다. 기저치와 치료 중 신기능을 평가하고, 신기능 장애 위험 인자에 유의하십시오. (5.14)
이상반응
가장 흔히 보고된 이상반응(30% 이상)은 부종, 구역질, 구토, 근육 경련, 근골격계 통증, 설사, 발진, 피로 및 복통입니다. (6.1)
의심되는 이상반응은 Amneal Pharmaceuticals 1-877-835-5472 또는 FDA 1-800-FDA-1088, www.fda.gov/medwatch로 보고하십시오.
약물상호작용
환자 상담 정보는 17항을 참조하십시오.
개정: 2023년 11월
목차
처방 정보 전문: 목차*
1 적응증 및 용법
1.1 새로 진단된 Philadelphia 양성 만성 골수성 백혈병(Ph+ CML)
1.2 Interferon-alpha(IFN) 치료 후 Blast Crisis(BC), Accelerated Phase(AP) 또는 Chronic Phase(CP)의 Ph+ CML
1.3 성인 Ph+ 급성 림프구성 백혈병(ALL) 환자
1.4 소아
Ph+ 급성 림프구성 백혈병(ALL) 환자
1.5 골수형성이상/골수증식성 질환(MDS/MPD)
1.6 공격적 전신성 Mastocytosis(ASM)
1.7 과호산구증후군(HES) 및/또는 만성 호산구성 백혈병(CEL)
1.8 Dermatofibrosarcoma Protuberans(DFSP)
1.9 Kit+ 위장관 기질종양(GIST)
1.10 GIST의
수술 후 보조요법
2 용법 및 투여방법
2.1 약물 투여
2.2 Ph+ CML CP, AP 또는 BC 성인 환자
2.3 Ph+ CML CP
소아 환자
2.4 Ph+ ALL 성인 환자
2.5
Ph+ ALL 소아 환자
2.6 MDS/MPD 성인 환자
2.7 ASM 성인 환자
2.8 HES/CEL 성인 환자
2.9 DFSP 성인 환자
2.10 전이성 및/또는 절제 불가능한 GIST 성인 환자
2.11 수술 후 보조요법 GIST 성인 환자
2.12 용량 조절 지침
2.13 간독성 및 비혈액학적 부작용에 대한 용량 조절
2.14 혈액학적 부작용에 대한 용량 조절
3 제형 및 함량
4 금기사항
5 경고 및 주의사항
5.1 체액 저류 및 부종
5.2 혈액학적 독성
5.3 울혈성 심부전 및 좌심실 기능장애
5.4 간독성
5.5 출혈
5.6 위장관계 장애
5.7 과호산구성 심장 독성
5.8 피부 독성
5.9 갑상선기능저하증
5.10 태아 독성
5.11 소아 및 청소년의 성장 지연
5.12 종양 용해 증후군
5.13 운전 및 기계 사용 능력 저하
5.14 신장 독성
6 부작용
6.1 임상시험 경험
6.2 시판 후 경험
7 약물 상호작용
7.1 CYP3A 대사를 유도하는 약물
7.2 CYP3A 대사를 억제하는 약물
7.3 CYP3A4에 의해 대사되는 약물과의 상호작용
7.4 CYP2D6에 의해 대사되는 약물과의 상호작용
8 특정 집단에서의 사용
8.1 임신
8.2 수유
8.3 가임기 여성 및 남성
8.4 소아 사용
8.5 노인 사용
8.6 간장애 환자
8.7 신장애 환자
10 과량투여
11 설명
12 임상약리
12.1 작용기전
12.3 약동학
13 비임상 독성
13.1 발암성, 변이원성, 생식능력 장애
13.2 동물 독성 및/또는 약리
14 임상시험
14.1 만성 골수성 백혈병
14.2 소아 CML
14.3 급성 림프구성 백혈병
14.4 소아 ALL
14.5 골수형성이상/골수증식성 질환
14.6 공격적 전신성 Mastocytosis
14.7 과호산구증후군/만성 호산구성 백혈병
14.8 Dermatofibrosarcoma Protuberans
14.9 위장관 기질종양
15 참고문헌
16 공급/보관 및 취급방법
17 환자 상담 정보
- *
- 처방 정보 전문에서 생략된 섹션 또는 하위 섹션은 나열되지 않았습니다.
1 적응증 및 사용법
1.1 새로 진단된 Philadelphia Positive Chronic Myeloid Leukemia (Ph+ CML)
새로 진단된 성인 및 소아 환자의 만성기 Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML).
1.2 Interferon-alpha (IFN) 치료 후 Blast Crisis (BC), Accelerated Phase (AP) 또는 Chronic Phase (CP)의 Ph+ CML
Blast crisis, accelerated phase 또는 interferon-alpha 치료 실패 후 만성기의 Philadelphia chromosome positive chronic myeloid leukemia 환자.
1.3 Ph+ Acute Lymphoblastic Leukemia (ALL)의 성인 환자
재발성 또는 불응성 Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL)의 성인 환자.
1.4 Ph+ Acute Lymphoblastic Leukemia (ALL)의 소아 환자
항암 화학요법과 병용한 새로 진단된 Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL)의 소아 환자.
1.5 Myelodysplastic/Myeloproliferative Diseases (MDS/MPD)
Platelet-derived growth factor receptor (PDGFR) 유전자 재배열이 있는 myelodysplastic/myeloproliferative diseases의 성인 환자.
1.6 Aggressive Systemic Mastocytosis (ASM)
D816V c-Kit 변이가 없거나 c-Kit 변이 상태가 알려지지 않은 aggressive systemic mastocytosis의 성인 환자.
1.7 Hypereosinophilic Syndrome (HES) 및/또는 Chronic Eosinophilic Leukemia (CEL)
FIP1L1-PDGFRα fusion kinase 양성(변이 분석 또는 CHIC2 allele 결실의 FISH 증명)인 hypereosinophilic syndrome 및/또는 chronic eosinophilic leukemia의 성인 환자 및 FIP1L1-PDGFRα fusion kinase 음성이거나 알려지지 않은 HES 및/또는 CEL 환자.
1.8 Dermatofibrosarcoma Protuberans (DFSP)
절제 불가능, 재발성 및/또는 전이성 dermatofibrosarcoma protuberans의 성인 환자.
1.9 Kit+ Gastrointestinal Stromal Tumors (GIST)
절제 불가능 및/또는 전이성 악성 gastrointestinal stromal tumors에서 Kit (CD117) 양성인 환자.
2 용량 및 투여
2.1 약물 투여
처방된 용량은 식사와 함께 큰 양의 물과 함께 경구로 투여되어야 합니다. 400 mg 또는 600 mg의 용량은 하루에 한 번 투여되어야 하며, 800 mg의 용량은 하루에 400 mg를 두 번 투여해야 합니다.
필름 코팅된 정제를 삼키지 못하는 환자의 경우, 정제를 물이나 사과 주스에 분산시킬 수 있습니다. 필요한 정제의 개수는 음료의 적절한 용량(100 mg 정제의 경우 약 50 mL, 400 mg 정제의 경우 약 200 mL)에 넣고 숟가락으로 저어야 합니다. 정제가 완전히 분해된 후에 즉시 액상을 투여해야 합니다.
800 mg 이상의 일일 투여량의 경우, 철분에 노출되지 않도록 400 mg 정제를 사용하여 투여해야 합니다.
진행성 질환이나 용인할 수 없는 독성이 없는 한 치료는 계속할 수 있습니다.
2.2 만성 기간 CML CP, AP 또는 BC를 가진 성인 환자
만성 기간 CML의 성인 환자에게는 imatinib mesylate 정제의 권장 용량은 하루에 400 mg이며, 가속 기간 또는 블라스트 위기의 성인 환자에게는 하루에 600 mg입니다.
CML에서 만성 기간 질환을 가진 성인 환자의 경우 400 mg에서 600 mg로 용량을 증가시킬 수 있으며, 가속 기간 또는 블라스트 위기의 성인 환자의 경우 심각한 약물 반응 및 중성구 감소증이 없는 경우 다음과 같은 상황에서 고려할 수 있습니다: 질환 진행(언제든지), 치료 후 최소 3개월 동안 만족스러운 혈액학적 반응을 얻지 못한 경우, 치료 후 6~12개월 동안 세포유전학적 반응을 얻지 못한 경우, 이전에 얻은 혈액학적 또는 세포유전학적 반응을 상실한 경우.
2.3 만성 기간 CML을 가진 소아 환자
신규 진단을 받은 만성 기간 CML을 가진 소아에게는 imatinib mesylate 정제의 권장 용량은 340 mg/m2/day(600 mg를 초과하지 않음)입니다. imatinib mesylate 정제 치료는 하루에 한 번 투여하거나 하루 용량을 두 번으로 나누어 아침에 한 부분을 투여하고 저녁에 한 부분을 투여할 수 있습니다. 1세 미만의 소아에서는 imatinib mesylate 정제 치료에 대한 경험이 없습니다.
2.4 재발/저항성 Ph+ ALL을 가진 성인 환자
재발/저항성 Ph+ ALL을 가진 성인 환자에게는 imatinib mesylate 정제의 권장 용량은 하루에 600 mg입니다.
2.5 신규 진단을 받은 Ph+ ALL을 가진 소아 환자
신규 진단을 받은 Ph+ ALL을 가진 소아에게는 화학 요법과 병용하여 imatinib mesylate 정제의 권장 용량은 340 mg/m2/day(600 mg를 초과하지 않음)입니다. imatinib mesylate 정제 치료는 하루에 한 번 투여될 수 있습니다.
2.6 MDS/MPD를 가진 성인 환자
치료를 시작하기 전에 PDGFRb 유전자 재배열 상태를 확인하십시오.
imatinib mesylate 정제의 권장 용량은 MDS/MPD를 가진 성인 환자에게 하루에 400 mg입니다.
2.7 ASM을 가진 성인 환자
치료를 시작하기 전에 D816V c-Kit 돌연변이 상태를 확인하십시오.
imatinib mesylate 정제의 권장 용량은 D816V c-Kit 돌연변이가 없는 ASM을 가진 성인 환자에게 하루에 400 mg입니다. c-Kit 돌연변이 상태가 알려지지 않거나 알 수 없는 경우, 다른 치료에 만족스럽게 반응하지 않는 ASM 환자에게 imatinib mesylate 정제 400 mg/day 치료를 고려할 수 있습니다. 과립구증과 관련된 클론 혈액학적 질환인 FIP1L1-PDGFRα 융합 키나제와 관련된 ASM 환자의 경우, 100 mg/day의 시작 용량이 권장됩니다. 이러한 환자의 경우 치료에 대한 충분한 반응이 없는 것으로 평가되면 약물 반응이 없는 경우 100 mg에서 400 mg로 용량을 증가시킬 수 있습니다.
2.8 HES/CEL을 가진 성인 환자
imatinib mesylate 정제의 권장 용량은 HES/CEL을 가진 성인 환자에게 하루에 400 mg입니다. FIP1L1-PDGFRα 융합 키나제가 확인된 HES/CEL 환자의 경우, 100 mg/day의 시작 용량이 권장됩니다. 이러한 환자의 경우 치료에 대한 충분한 반응이 없는 것으로 평가되면 약물 반응이 없는 경우 100 mg에서 400 mg로 용량을 증가시킬 수 있습니다.
2.10 전이성 및/또는 절제 불가능한 GIST를 가진 성인 환자
imatinib mesylate 정제의 권장 용량은 전이성 및/또는 절제 불가능한 악성 GIST를 가진 성인 환자에게 하루에 400 mg입니다. 낮은 용량에서 질병 진행의 명확한 증상이나 징후가 나타나는 환자에서는 필요에 따라 최대 800 mg까지 용량을 증가시킬 수 있습니다. 이 경우 심각한 약물 반응이 없는 경우에만 증량해야 합니다.
2.11 GIST의 보조 치료를 받는 성인 환자
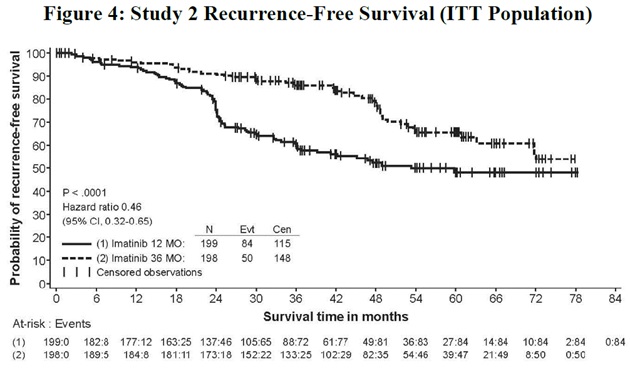
imatinib mesylate 정제의 권장 용량은 GIST의 전적인 총 절제 후 성인 환자의 보조 치료를 위해 하루에 400 mg입니다. 임상 시험에서는 imatinib mesylate 정제 1년과 imatinib mesylate 정제 3년이 연구되었습니다. 연구 2에서 정의된 환자 인구에서는 imatinib mesylate 정제 3년이 권장됩니다 [자세한 내용은 임상 연구 (14.8) 참조]. imatinib mesylate 정제의 최적 치료 기간은 알려져 있지 않습니다.
2.12 용량 변경 가이드라인
강력한 CYP3A4 유도제와의 병용 투여: 강력한 CYP3A4 유도제(예, dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, rifampacin, phenobarbital)와의 병용 투여는 피해야 한다. 환자가 반드시 강력한 CYP3A4 유도제를 병용 투여해야 하는 경우, 약동학 연구에 근거하여 imatinib mesylate 정제의 용량을 최소 50% 증량해야 하며, 임상 반응을 주의 깊게 모니터링해야 한다 [Drug Interactions (7.1) 참조].
간 장애: 경증 및 중등도의 간 장애 환자는 용량 조절이 필요하지 않으며 권장 용량으로 치료해야 한다. 중증의 간 장애 환자의 경우 권장 용량의 25%를 감량해야 한다 [Use in Specific Populations (8.6) 참조].
신장 장애: 중등도의 신장 장애(creatinine clearance [CrCL] = 20 to 39 mL/min) 환자는 권장 초기 용량을 50% 감량해야 하며 이후 용량은 내약성에 따라 증량할 수 있다. 경증의 신장 장애(CrCL = 40 to 59 mL/min) 환자에게는 600 mg을 초과하는 용량이 권장되지 않는다. 중등도의 신장 장애 환자의 경우 400 mg을 초과하는 용량은 권장되지 않는다.
중증의 신장 장애 환자에서는 주의해서 imatinib을 사용해야 한다. 중증의 신장 장애 환자 2명에서 100 mg/day 용량이 내약성을 보였다 [Warnings and Precautions (5.3), Use in Specific Populations (8.7) 참조].
2.13 간독성 및 비혈액학적 이상반응에 대한 용량 조절
빌리루빈이 기관 정상 상한치(IULN)의 3배를 초과하거나 간 transaminase가 IULN의 5배를 초과하는 상승이 발생하면, 빌리루빈 수치가 IULN의 1.5배 미만으로 회복되고 transaminase 수치가 IULN의 2.5배 미만으로 회복될 때까지 imatinib mesylate 정제를 중단해야 한다. 성인의 경우, imatinib mesylate 정제를 감량된 일일 용량(예, 400 mg에서 300 mg으로, 600 mg에서 400 mg으로 또는 800 mg에서 600 mg으로)으로 치료를 지속할 수 있다. 소아에서는 340 mg/m2/day에서 260 mg/m2/day로 같은 상황에서 일일 용량을 감량할 수 있다.
중증의 비혈액학적 이상반응(예, 중증 간독성 또는 중증 체액 저류)이 발생하는 경우, 이상반응이 해결될 때까지 imatinib mesylate 정제를 중단해야 한다. 이후, 이상반응의 초기 중증도에 따라 적절하게 치료를 재개할 수 있다.
2.14 혈액학적 이상반응에 대한 용량 조절
중증 호중구 감소증 및 혈소판 감소증에 대해서는 Table 1에 제시된 대로 용량 감량 또는 치료 중단이 권장된다.
표 1: 호중구 감소증 및 혈소판 감소증에 대한 용량 조절
|
호산구증을 동반한 ASM |
ANC 1.0 x 109/L 미만 |
1. ANC가 1.5 x 109/L 이상이 되고 혈소판이 75 x 109/L 이상이 될 때까지 imatinib mesylate 정제 투여를 중단한다 2. 이전 용량(즉, 중증 이상반응 발생 전 용량)으로 imatinib mesylate 정제 치료를 재개한다 |
|
FIP1L1-PDGFRα fusion kinase를 동반한 HES/CEL(초기 용량 100 mg) |
ANC 1.0 x 109/L 미만 |
1. ANC가 1.5 x 109/L 이상이 되고 혈소판이 75 x 109/L 이상이 될 때까지 imatinib mesylate 정제 투여를 중단한다 2. 이전 용량(즉, 중증 이상반응 발생 전 용량)으로 imatinib mesylate 정제 치료를 재개한다 |
|
만성기 CML(초기 용량 400 mg) MDS/MPD, ASM 및 HES/CEL GIST(초기 용량 400 mg)
|
ANC 1.0 x 109/L 미만 |
1. ANC가 1.5 x 109/L 이상이 되고 혈소판이 75 x 109/L 이상이 될 때까지 imatinib mesylate 정제 투여를 중단한다 2. 초기 시작 용량인 400 mg으로 imatinib mesylate 정제 치료를 재개한다 3. ANC 1.0 x 109/L 미만 및/또는 혈소판 50 x 109/L 미만이 재발하면, 1단계를 반복하고 imatinib mesylate 정제를 300 mg으로 감량하여 재개한다 |
|
Ph+ CML: 가속기 및 (초기 용량 600 mg) |
ANC 0.5 x 109/L 미만 |
1. 혈구 감소증이 백혈병과 관련이 있는지 확인한다(골수 흡인 또는 생검) 2. 혈구 감소증이 백혈병과 무관한 경우, imatinib mesylate 정제 용량을 400 mg으로 감량한다 3. 혈구 감소증이 2주 지속되면 300 mg으로 추가 감량한다 4. 혈구 감소증이 4주 지속되고 여전히 백혈병과 무관한 경우, ANC가 1 x 109/L 이상이 되고 혈소판이 20 x 109/L 이상이 될 때까지 imatinib mesylate 정제 투여를 중단하고 300 mg으로 치료를 재개한다 |
|
DFSP |
ANC 1.0 x 109/L 미만 |
1. ANC가 1.5 x 109/L 이상이 되고 혈소판이 75 x 109/L 이상이 될 때까지 imatinib mesylate 정제 투여를 중단한다 2. 600 mg으로 imatinib mesylate 정제 치료를 재개한다 3. 절대 호중구 수가 1.0 x 109/L 미만이고/또는 혈소판 수치가 50 x 109/L 미만으로 재발한 경우, 1단계를 반복하고 이마티닙 메실레이트 정제를 400mg의 감량된 용량으로 재투여합니다. |
|
소아 신규 만성기 |
절대 호중구 수 1.0 x 109/L 미만 |
1. 절대 호중구 수가 1.5 x 109/L 이상, 혈소판 수가 75 x 109/L 이상이 될 때까지 이마티닙 메실레이트 정제 투여를 중단합니다. 2. 이전 용량(즉, 중증 이상반응 전 용량)으로 이마티닙 메실레이트 정제 투여를 재개합니다. 3. 절대 호중구 수가 1.0 x 109/L 미만이고/또는 혈소판 수치가 50 x 109/L 미만으로 재발한 경우, 1단계를 반복하고 이마티닙 메실레이트 정제를 260mg/m2의 감량된 용량으로 재투여합니다. |
|
약어: ANC, 절대 호중구 수; ASM, 공격적 전신성 비만세포증; CEL, 만성 호산구성 백혈병; CML, 만성 골수성 백혈병; DFSP, 피부 섬유육종 돌출종; HES, 과잉 호산구 증후군; MDS/MPD, 골수증식성질환/골수이형성질환; PDGFR, 혈소판 유래 성장인자 수용체; Ph+ CML, 필라델피아 염색체 양성 만성 골수성 백혈병; Ph+ ALL, 필라델피아 염색체 양성 급성 림프구성 백혈병. |
||
3 제형 및 함량
100 mg 필름코팅정:
갈색, 원형, 분할선이 있는 필름코팅정으로, 분할선 측면에는 “AN”이, 반대 측면에는 “794”가 각인되어 있습니다.
400 mg 필름코팅정:
갈색, 타원형, 분할선이 있는 필름코팅정으로, 분할선 측면에는 “AN”이, 반대 측면에는 “795”가 각인되어 있습니다.
4 금기사항
없음.
5 경고 및 주의사항
5.1 체액 저장 및 부종
이마티닙 메실레이트는 종종 부종과 가끔 심각한 체액 저장과 관련이 있습니다 [자세한 내용은 부작용 (6.1)을 참조하세요]. 체액 저장의 징후와 증상을 정기적으로 체중을 측정하고 모니터링하세요. 예기치 않은 급격한 체중 증가를 주의 깊게 조사하고 적절한 치료를 제공하세요. CML 연구에서 이마티닙 메실레이트 용량이 높고 65세 이상의 연령에서 부종의 가능성이 증가했습니다. 신규 진단 CML 환자 중 1.5%가 이마티닙 메실레이트를 복용하고 있을 때 심한 표피 부종이 보고되었으며, 다른 성인 CML 환자 중 2%에서 6%가 이마티닙 메실레이트를 복용하고 있을 때 심한 체액 저장(예: 흉막 삼출, 심낭 삼출, 폐부종 및 복수) 반응이 보고되었습니다. GIST를 위해 이마티닙 메실레이트를 복용하는 환자 중 9%에서 13.1%가 심한 체액 저장이 보고되었습니다 [자세한 내용은 부작용 (6.1)을 참조하세요]. 만성 위암상 CML 환자를 대상으로 한 무작위 임상시험에서 이마티닙 메실레이트와 니로티닙을 비교한 결과, 심한(3 또는 4급) 체액 저장이 이마티닙 메실레이트를 복용한 환자 중 2.5%에서 발생하였으며, 니로티닙 300mg를 하루에 두 번 복용한 환자 중 3.9%에서 발생하였습니다. 이마티닙 메실레이트 그룹에서는 2.1%의 환자(3 또는 4급은 없음)에서 삼출(흉막 삼출, 심낭 삼출, 복수) 또는 폐부종이 관찰되었으며, 니로티닙 300mg를 하루에 두 번 복용한 그룹에서는 2.2%의 환자(0.7%가 3 또는 4급)에서 관찰되었습니다.
5.2 혈액 독성
이마티닙 메실레이트 치료는 빈혈, 중성구 감소 및 혈소판 감소와 관련이 있습니다. 처음 한 달 동안은 매주 완전한 혈액 검사를 하고, 두 번째 달에는 2주마다, 그 후에는 임상적으로 필요한 경우(예: 2~3개월마다) 주기적으로 혈액 검사를 수행하세요. CML에서 이러한 세포 감소는 질병의 단계에 따라 다르며, 만성 위암상 CML 환자보다 가속 위암상 CML 또는 블라스트 위기 환자에서 더 빈번하게 발생합니다. 소아 CML 환자에서 가장 빈번하게 관찰되는 독성은 중성구 감소, 혈소판 감소 및 빈혈을 포함한 3 또는 4급의 세포 감소입니다. 이러한 증상은 일반적으로 치료 시작 후 처음 몇 개월 내에 발생합니다 [자세한 내용은 용량 및 투여 방법 (2.14)을 참조하세요].
5.3 심부전 및 좌심실 기능 장애
이마티닙 메실레이트를 복용하는 환자들에서 심부전 및 좌심실 기능 장애가 보고되었습니다. 심장 부작용은 고령자나 과거 심장 질환의 의료 기록을 가진 환자에서 더 빈번하게 발생합니다. 만성 위암상 CML 환자를 대상으로 한 국제적인 무작위 임상 3상 연구에서 이마티닙 메실레이트를 복용한 환자 중 0.7%에서 심한 심장 부전 및 좌심실 기능 장애가 관찰되었으며, IFN + Ara-C를 복용한 환자 중 0.9%에서 관찰되었습니다. 이마티닙 메실레이트와 니로티닙을 비교한 다른 임상시험에서도 만성 위암상 CML 환자를 대상으로 한 신규 진단 연구에서 심부전이 이마티닙 메실레이트를 복용한 환자 중 1.1%에서 관찰되었으며, 니로티닙 300mg를 하루에 두 번 복용한 환자 중 2.2%에서 관찰되었으며, 심한(3 또는 4급) 심부전은 양쪽 그룹에서 각각 0.7%의 환자에서 발생하였습니다. 심장 질환 또는 심장 질환 위험 요인 또는 신장 부전의 병력이 있는 환자를 주의 깊게 모니터링하세요. 심장 또는 신장 부전과 일치하는 증상이나 징후가 있는 환자를 평가하고 치료하세요.
5.4 간독성
이마티닙 메실레이트 복용 시 간독성이 가끔 심각할 수 있습니다 [자세한 내용은 부작용 (6.1)을 참조하세요]. 단기 또는 장기간 이마티닙 메실레이트 사용 시 치명적인 간부전 및 심한 간손상으로 이어질 수 있습니다. 치료 시작 전과 월간 또는 임상적으로 필요한 경우 간 기능 검사(전이아미나제, 빌리루빈 및 알칼리성 인산효소)를 모니터링하세요. 이마티닙 메실레이트 중단 및/또는 용량 감소로 실험실 이상을 관리하세요 [자세한 내용은 용량 및 투여 방법 (2.13)을 참조하세요]. 이마티닙 메실레이트와 화학요법을 병용한 경우, 아미노전이효소 상승 및 고빌리루빈혈증 형태로 간 독성이 관찰되었습니다. 또한 급성 간부전의 보고가 있었습니다. 간 기능 모니터링이 권장됩니다.
5.5 출혈
신규 진단 CML 환자를 대상으로 한 이마티닙 메실레이트 대 IFN+Ara-C 임상시험에서 1.8%의 환자가 3/4급 출혈을 경험했습니다. 3상 불수술적 또는 전이성 GIST 연구에서 211명의 환자(12.9%)가 어떤 부위에서든 3/4급 출혈을 보고했습니다. 2상 불수술적 또는 전이성 GIST 연구에서 7명의 환자(5%)가 총 8건의 CTC 3/4급 출혈을 경험했습니다. 위장관(3명), 종양 내(3명) 또는 둘 다(1명) 출혈이 있었습니다. 위장관 종양 부위가 위장관 출혈의 원인이 될 수 있습니다. 만성 위암상 CML 환자를 대상으로 한 무작위 임상시험에서 이마티닙 메실레이트 그룹에서 위장관 출혈이 1.4%의 환자에서 관찰되었으며, 니로티닙 300mg를 하루에 두 번 복용한 그룹에서는 2.9%의 환자에서 관찰되었습니다. 이마티닙 메실레이트 그룹에서는 이러한 사건 중 3/4급은 없었으며, 니로티닙 300mg를 하루에 두 번 복용한 그룹에서는 0.7%가 3/4급이었습니다. 또한 시판 후 경험에서 위의 혈관 이형성증이 보고되었습니다.
5.6 위장 장애
이마티닙 메실레이트는 때로 위장관 자극과 관련이 있습니다. 이마티닙 메실레이트는 음식과 함께 큰 양의 물과 함께 복용하여 이 문제를 최소화해야 합니다. 위장관 천공을 포함한 드문 보고가 있었습니다.
5.7 과다한 호산구 심장 독성
심근 내 HES 세포의 잠복 침윤을 가진 과다한 호산구증후군 환자에서 이마티닙 메실레이트 치료 시작 시 HES 세포의 변성에 따른 심근성 쇼크/좌심실 기능 장애가 관찰되었습니다. 이 상태는 전신 스테로이드 투여, 순환 지원 조치 및 일시적으로 이마티닙 메실레이트를 중단함으로써 회복되었습니다.
고백형성장장애/혈구증가성질환 및 전신마스토세토시스는 고이소구증과 관련될 수 있습니다. HES/CEL 환자와 고이소구증과 관련된 MDS/MPD 또는 ASM 환자에게 심장초음파 및 혈청트로포닌 검사를 고려하십시오. 어느 하나라도 비정상적인 경우, 치료 시작 시 imatinib mesylate와 동시에 1~2mg/kg의 시스테믹 스테로이드를 1~2주간 예방적으로 사용하는 것을 고려하십시오.
5.8 피부독성
imatinib mesylate 사용과 관련하여 홍반성 다양한 피부반응 및 스티븐스-존슨 증후군을 포함한 불루스 피부반응이 보고되었습니다. 홍반성 다양한 피부반응, 스티븐스-존슨 증후군을 포함한 불루스 피부반응의 경우, 후처치 중 재발하는 피부반응이 관찰되었습니다. 해외 후처치 보고서에서는 반응이 해결되거나 개선된 후 imatinib mesylate 치료를 다시 시작한 환자들이 있었습니다. 이러한 경우, imatinib mesylate는 반응이 발생한 용량보다 낮은 용량으로 재개되었으며 일부 환자는 스테로이드나 항히스타민제와 함께 병용 치료를 받았습니다.
5.9 갑상선기능저하증
갑상선절제술을 받은 환자에서 imatinib mesylate 치료 중 갑상선기능저하증의 임상적 사례가 보고되었습니다. 이러한 환자들의 TSH 수치를 모니터링하십시오.
5.10 태아-배아 독성
임신한 여성에게 imatinib mesylate를 투여할 경우 태아에 유해를 일으킬 수 있습니다. imatinib mesylate는 장기형성기에 쥐에서 태아기에 투여할 경우 최대 인간용량인 800mg/일에 해당하는 용량에서 기형을 유발했습니다. 쥐에게 최대 인간용량의 절반에 해당하는 용량으로 imatinib mesylate를 투여한 경우, 중대한 이식 후 손실이 관찰되었습니다. 생식력이 있는 여성 환자에게 imatinib mesylate를 사용할 때 효과적인 피임법(임신율이 1% 미만인 방법)을 사용하도록 권장하고, imatinib mesylate 사용 후 14일 동안 효과적인 피임법을 사용하도록 안내하십시오. 이 약물을 임신 중에 사용하거나 환자가 이 약물을 복용하는 동안 임신할 경우 태아에 대한 잠재적인 위험에 대해 환자에게 알리십시오 [자세한 내용은 특정 인구에서의 사용 (8.1) 참조].
5.11 소아 및 청소년의 성장지연
imatinib mesylate를 복용하는 소아 및 사전 청소년기 아동에서 성장지연이 보고되었습니다. 소아에서 imatinib mesylate를 장기간 투여하는 것이 성장에 미치는 장기적인 영향은 알려지지 않았습니다. 따라서 imatinib mesylate 치료 중인 소아의 성장을 모니터링하십시오 [부작용 (6.1) 참조].
5.12 종양용해증후군
CML, GIST, ALL 및 고이소구증 환자에서 imatinib mesylate를 복용하는 동안 종양용해증후군(TLS) 사례, 치명적인 사례를 포함하여 보고되었습니다. TLS 위험을 가진 환자는 치료 전 종양의 고증식률 또는 고종양부하를 가진 환자입니다. 이러한 환자들을 밀접하게 모니터링하고 적절한 예방 조치를 취하십시오. TLS의 가능성으로 인해, 임박한 임상적으로 중요한 탈수와 높은 요산 수치를 치료하기 전에 imatinib mesylate 치료를 시작하십시오.
5.13 운전 및 기계 사용과 관련된 장애
imatinib mesylate를 복용하는 환자들에서 자동차 사고가 보고되었습니다. 환자들에게 어지러움, 시야 흐림 또는 졸음 등의 부작용이 발생할 수 있으므로 imatinib mesylate 치료 중에는 운전 및 기계 작동 시 주의를 요하도록 권고하십시오.
5.14 신장 독성
imatinib mesylate를 복용하는 환자들에서 신장 기능 저하가 발생할 수 있습니다. 새로 진단받은 CML 환자(4개의 무작위화 임상시험) 및 악성 GIST 환자(1개의 단일암 임상시험)에서 imatinib mesylate 400mg를 매일 복용한 환자들의 중앙 추정 사구체 여과율(eGFR) 값은 기준치인 85 mL/min/1.73m2(N=1,190)에서 12개월 후 75 mL/min/1.73m2(N=1,082)로 감소하였으며 60개월 후 69 mL/min/1.73m2(N=549)로 감소하였습니다. imatinib mesylate 치료 시작 전에 신장 기능을 평가하고, 신장 기능에 대한 위험 요소(기존 신장 손상, 당뇨병, 고혈압 및 심부전 등)에 주의하여 치료 중에 신장 기능을 모니터링하십시오.
6 ADVERSE REACTIONS
다음의 중대한 이상반응은 라벨의 다른 부분에서 설명되어 있습니다.
- 체액 저류 및 부종 [경고 및 주의사항(5.1) 참조]
- 혈액학적 독성 [경고 및 주의사항(5.2) 참조]
- 울혈성 심부전 및 좌심실 기능 장애 [경고 및 주의사항(5.3) 참조]
- 간독성 [경고 및 주의사항(5.4) 참조]
- 출혈 [경고 및 주의사항(5.5) 참조]
- 위장관 장애 [경고 및 주의사항(5.6) 참조]
- 호산구증가증 심장 독성 [경고 및 주의사항(5.7) 참조]
- 피부 독성 [경고 및 주의사항(5.8) 참조]
- 갑상선기능저하증 [경고 및 주의사항(5.9) 참조]
- 소아 및 청소년의 성장 지연 [경고 및 주의사항(5.11) 참조]
- 종양 용해 증후군 [경고 및 주의사항(5.12) 참조]
- 운전 및 기계 조작 능력 저하 [경고 및 주의사항(5.13) 참조]
- 신장 독성 [경고 및 주의사항(5.14) 참조]
6.1 임상시험 경험
임상시험은 광범위하게 다양한 조건에서 실시되므로 임상시험에서 관찰된 이상반응 발생률을 다른 약물의 임상시험에서 관찰된 발생률과 직접 비교할 수 없으며, 실제 환자에서 관찰되는 발생률을 반영하지 않을 수 있습니다.
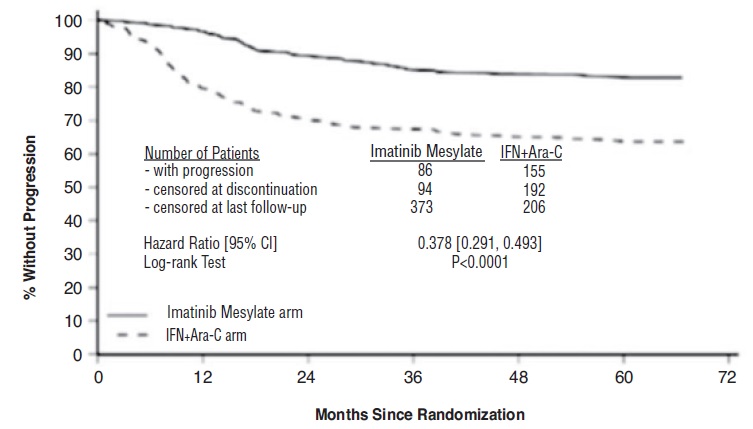
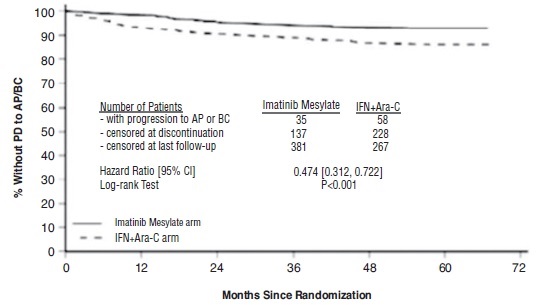
만성 골수성 백혈병
이마티닙메실레이트로 치료받은 대부분의 환자들은 언젠가 이상반응을 경험했습니다. Ph+ CML 만성기 신규 진단 환자를 대상으로 이마티닙메실레이트 대 IFN+Ara-C를 비교한 무작위 배정 임상시험에서 이마티닙메실레이트로 인한 이상반응으로 투여를 중단한 경우는 2.4%였고, Ph+ CML 만성기 신규 진단 환자를 대상으로 이마티닙메실레이트와 닐로티닙을 비교한 무작위 배정 임상시험에서는 12.5%였습니다. 인터페론-알파 치료에 실패한 만성기 환자에서 약물 관련 이상반응으로 투여를 중단한 경우는 4%, 가속기에서는 4%, 급성기에서는 5%였습니다.
가장 빈번하게 보고된 약물 관련 이상반응은 부종, 구역 및 구토, 근육 경련, 근골격계 통증, 설사 및 발진이었습니다(표 2 및 표 3은 신규 진단 CML, 표 4는 기타 CML 환자). 부종은 주로 안와 주위 또는 하지에서 발생했으며, 이뇨제, 기타 보조 요법 또는 이마티닙메실레이트의 용량 감량으로 관리되었습니다[투여량 및 투여방법(2.13) 참조]. 중증 표재성 부종의 발생 빈도는 1.5% ~ 6%였습니다.
다양한 이상반응은 국소 또는 전신 체액 저류를 나타냅니다. 여기에는 흉막 삼출, 복수, 폐부종 및 표재성 부종이 있거나 없는 급격한 체중 증가가 포함됩니다. 이러한 반응은 용량 의존적인 것으로 보이며, 급성기 및 가속기 연구(용량이 600 mg/일이었음)에서 더 흔했고, 고령자에서 더 흔한 것으로 나타났습니다. 이러한 반응은 일반적으로 이마티닙메실레이트 투여를 중단하고 이뇨제 또는 기타 적절한 보조 처치를 사용하여 관리되었습니다. 이러한 반응은 중대하거나 생명을 위협할 수 있습니다.
시험 약물과 관계없이 이마티닙메실레이트로 치료받은 환자에서 10% 이상 보고된 이상반응은 표 2, 3 및 4에 나와 있습니다.
표 2: IFN+Ara-C 대비 이마티닙메실레이트 신규 진단 CML 임상시험에서 보고된 시험약물과의 관련성 여부에 관계없이 10% 이상 발생한 이상반응(1)
|
전체 등급 |
CTC 등급* 3/4 |
|||
|
선호 용어 |
이마티닙메실레이트 |
IFN+Ara−C |
이마티닙메실레이트 |
IFN+Ara−C |
|
체액 저류 |
61.7 |
11.1 |
2.5 |
0.9 |
|
− 표재성 부종 |
59.9 |
9.6 |
1.5 |
0.4 |
|
− 기타 체액 저류 반응2 |
6.9 |
1.9 |
1.3 |
0.6 |
|
구역 |
49.5 |
61.5 |
1.3 |
5.1 |
|
근육 경련 |
49.2 |
11.8 |
2.2 |
0.2 |
|
근골격 통증 |
47.0 |
44.8 |
5.4 |
8.6 |
|
설사 |
45.4 |
43.3 |
3.3 |
3.2 |
|
발진 및 관련 용어 |
40.1 |
26.1 |
2.9 |
2.4 |
|
피로 |
38.8 |
67.0 |
1.8 |
25.1 |
|
두통 |
37.0 |
43.3 |
0.5 |
3.8 |
|
관절 통증 |
31.4 |
38.1 |
2.5 |
7.7 |
|
복통 |
36.5 |
25.9 |
4.2 |
3.9 |
|
비인두염 |
30.5 |
8.8 |
0 |
0.4 |
|
출혈 |
28.9 |
21.2 |
1.8 |
1.7 |
|
– 위장출혈 |
1.6 |
1.1 |
0.5 |
0.2 |
|
– 중추신경계 출혈 |
0.2 |
0.4 |
0 |
0.4 |
|
근통 |
24.1 |
38.8 |
1.5 |
8.3 |
|
구토 |
22.5 |
27.8 |
2.0 |
3.4 |
|
소화불량 |
18.9 |
8.3 |
0 |
0.8 |
|
기침 |
20.0 |
23.1 |
0.2 |
0.6 |
|
인두통 |
18.1 |
11.4 |
0.2 |
0 |
|
상부 호흡기 감염 |
21.2 |
8.4 |
0.2 |
0.4 |
|
어지러움 |
19.4 |
24.4 |
0.9 |
3.8 |
|
열 |
17.8 |
42.6 |
0.9 |
3.0 |
|
체중 증가 |
15.6 |
2.6 |
2.0 |
14.7 |
18.6 |
0 |
2.3 |
|
우울증 |
14.9 |
35.8 |
0.5 |
13.1 |
|
독감 |
13.8 |
6.2 |
0.2 |
0.2 |
|
뼈 통증 |
11.3 |
15.6 |
1.6 |
3.4 |
|
변비 |
11.4 |
14.4 |
0.7 |
0.2 |
|
부비동염 |
11.4 |
6.0 |
0.2 |
0.2 |
|
약어: CML, 만성 골수성 백혈병; CNS, 중추신경계; CTC, 일반용어기준; GI, 위장관; IFN, 인터페론-알파. * NCI 일반용어기준 이상반응, 버전 3.0. (1)이마티닙메실레이트 치료 환자의 10% 이상에서 발생한 모든 이상반응은 치료와의 의심되는 관련성에 관계없이 나열되었습니다. (2)기타 체액 저류 반응에는 흉막 삼출, 복수, 폐부종, 심낭 삼출, 전신부종, 악화된 부종 및 달리 명시되지 않은 체액 저류가 포함됩니다. |
||||
표 3: 새로 진단된 Ph+ CML-CP 환자에서 이마티닙메실레이트 대 닐로티닙 연구의 60개월 분석에서 가장 흔히 보고된 비혈액학적 이상반응(이마티닙메실레이트 400 mg 1일 1회 또는 닐로티닙 300 mg 1일 2회 그룹에서 10% 이상)a
|
새로 진단된 Ph+ CML-CP 환자 |
|||||
|
이마티닙메실레이트 400 mg 1일 1회 N=280 |
닐로티닙 300 mg 1일 2회 N=279 |
이마티닙메실레이트 400 mg 1일 1회 N=280 |
닐로티닙 300 mg 1일 2회 N=279 |
||
|
신체 부위 및 선호 용어 |
모든 등급 (%) |
CTC 등급b 3/4 (%) |
|||
|
피부 및 피하 조직 장애 |
발진 |
19 |
38 |
2 |
<1 |
|
가려움증 |
7 |
21 |
0 |
<1 |
|
|
탈모증 |
7 |
13 |
0 |
0 |
|
|
건조한 피부 |
6 |
12 |
0 |
0 |
|
|
위장관 장애 |
구역질 |
41 |
22 |
2 |
2 |
|
변비 |
8 6 부작용 |
20 |
0 |
<1 |
|
|
설사 |
46 |
19 |
4 |
1 |
|
|
구토 |
27 |
15 |
<1 |
<1 |
|
|
상복부 통증 |
14 |
18 |
<1 |
1 |
|
|
복통 |
12 |
15 |
0 |
2 |
|
|
소화불량 |
12 |
10 |
0 |
0 |
|
|
신경계 장애 |
두통 |
23 |
32 |
<1 |
3 |
|
현기증 |
11 |
12 |
<1 |
<1 |
|
|
전신 장애 및 투여 부위 상태 |
피로 |
20 |
23 |
1 |
1 |
|
발열 |
13 |
14 |
0 |
<1 |
|
|
권태감 |
12 |
14 |
0 |
<1 |
|
|
말초 부종 |
20 |
9 |
0 |
<1 |
|
|
안면 부종 |
14 |
<1 |
<1 |
0 |
|
|
근골격계 및 결합 조직 장애 |
근육통 |
19 |
19 |
<1 |
<1 |
|
관절통 |
17 |
22 |
<1 |
<1 |
|
|
근육 경련 |
34 |
12 |
1 |
0 |
|
|
사지 통증 |
16 |
15 |
<1 |
<1 |
|
|
요통 |
17 |
19 |
1 |
1 |
|
|
호흡기, 흉부 및 종격 장애 |
기침 |
13 |
17 6 이상반응 |
0 |
0 |
|
구인두 통증 |
6 |
12 |
0 |
0 |
|
|
호흡곤란 |
6 |
11 |
<1 |
2 |
|
|
감염 및 기생충 감염 |
비인두염 |
21 |
27 |
0 |
0 |
|
상기도 감염 |
14 |
17 |
0 |
<1 |
|
|
인플루엔자 |
9 |
13 |
0 |
0 |
|
|
위장염 |
10 |
7 |
<1 |
0 |
|
|
눈 장애 |
안검 부종 |
19 |
1 |
<1 |
0 |
|
안와주위 부종 |
15 |
<1 |
0 |
0 |
|
|
정신 장애 |
불면증 |
9 |
11 |
0 |
0 |
|
혈관 장애 |
고혈압 |
4 |
10 |
<1 |
1 |
|
약어: Ph+ CML-CP, 필라델피아 염색체 양성 만성 골수성 백혈병-만성기. a실험실 검사치 이상은 제외함. bNCI 일반 독성 기준, 버전 3.0. |
|||||
표 4: 다른 CML 임상시험에서 보고된 약물 관련 여부와 관계없는 이상반응(모든 시험에서 10% 이상 발생)(1)
|
골수모세포성 백혈병 |
급성전환기 |
만성기, IFN 실패 |
||||
|
이상반응 용어 |
모든 등급 |
3/4 등급 |
모든 등급 |
3/4 등급 |
모든 등급 |
3/4 등급 |
|
체액 저류 |
72 |
11 |
76 |
6 |
69 |
4 |
|
– 표층 부종 |
66 |
6 |
74 |
3 |
67 |
2 |
|
– 기타 체액 저류 6번 부작용 |
||||||
|
반응(2) |
22 |
6 |
15 |
4 |
7 |
2 |
|
구역질 |
71 |
5 |
73 |
5 |
63 |
3 |
|
근육 경련 |
28 |
1 |
47 |
0.4 |
62 |
2 |
|
구토 |
54 |
4 |
58 |
3 |
36 |
2 |
|
설사 |
43 |
4 |
57 |
5 |
48 |
3 |
|
출혈 |
53 |
19 |
49 |
11 |
30 |
2 |
|
– 중추신경계 출혈 |
9 |
7 |
3 |
3 |
2 |
1 |
|
– 위장관 출혈 |
8 |
4 |
6 |
5 |
2 |
0.4 |
|
근골격계 통증 |
42 |
9 |
49 |
9 |
38 |
2 |
|
피로 |
30 |
4 |
46 |
4 |
48 |
1 |
|
피부 발진 |
36 |
5 |
47 |
5 |
47 |
3 |
|
발열 |
41 |
7 |
41 |
8 |
21 |
2 |
|
관절통 |
25 |
5 |
34 |
6 |
40 |
1 |
|
두통 |
27 |
5 |
32 |
2 |
36 |
0.6 |
|
복통 |
30 |
6 |
33 |
4 |
32 |
|
|
몸무게 증가 |
5 |
1 |
17 |
5 |
32 |
7 |
|
기침 |
14 |
0.8 |
27 |
0.9 |
20 |
0 |
|
소화불량 |
12 |
0 |
22 |
0 |
27 |
0 |
|
근육통 |
9 |
0 |
24 |
2 |
27 |
0.2 |
|
코인두염 |
10 |
0 |
17 |
0 |
22 |
0.2 |
|
권태감 |
18 |
5 |
21 |
5 |
15 |
0.2 |
|
호흡곤란 |
15 |
4 |
21 |
7 |
12 |
0.9 |
|
상기도 감염 |
||||||
|
식욕부진 |
14 |
2 |
17 |
2 |
7 |
0 |
|
야간 발한 |
13 |
0.8 |
17 |
1 |
14 |
0.2 |
|
변비 |
16 |
2 |
16 |
0.9 |
9 |
0.4 |
|
현기증 |
12 |
0.4 |
13 |
0 |
16 |
0.2 |
|
인후염 |
10 |
0 |
12 |
0 |
15 |
0 |
|
불면증 |
10 |
0 |
14 |
0 |
14 |
0.2 |
|
가려움증 |
8 |
1 |
14 6번 부작용 |
0.9 |
14 |
0.8 |
|
저칼륨혈증 |
13 |
4 |
9 |
2 |
6 |
0.8 |
|
폐렴 |
13 |
7 |
10 |
7 |
4 |
1 |
|
불안 |
8 |
0.8 |
12 |
0 |
8 |
0.4 |
|
간 독성 |
10 |
5 |
12 |
6 |
6 |
3 |
|
Rigors |
10 |
0 |
12 |
0.4 |
10 |
0 |
|
가슴 통증 |
7 |
2 |
10 |
0.4 |
11 |
0.8 |
|
독감 |
0.8 |
0.4 |
6 |
0 |
11 |
0.2 |
|
부비동염 |
4 |
0.4 |
11 |
0.4 |
9 |
0.4 |
|
약어: CML, 만성골수성백혈병; IFN, 인터페론-알파. (1) 발생 비율이 10% 이상인 모든 부작용을 치료와의 관련성에 관계없이 나열하였습니다. (2) 기타 체액 저류 반응에는 흉막삼출, 복수, 폐부종, 심낭삼출, 전신부종, 악화된 부종, 체액저류(별도로 명시되지 않음) 등이 포함됩니다. |
||||||
혈액 및 생화학 검사 이상
특히 호중구감소증 및 혈소판감소증 등의 혈구감소증은 모든 연구에서 일관되게 나타났으며, 750mg 이상 용량에서 더 높은 빈도로 발생했습니다(1상 연구 결과). CML 환자에서의 혈구감소증 발생은 질병 단계에도 의존적이었습니다.
새로 진단된 CML 환자에서는 다른 CML 환자군에 비해 혈구감소증이 덜 자주 발생했습니다(표 5, 6, 7 참조). 3 또는 4등급의 호중구감소증 및 혈소판감소증의 빈도는 급성기 및 가속기 단계에서 만성기보다 2~3배 더 높았습니다(표 4, 5 참조). 호중구감소증 및 혈소판감소증의 중간 지속 기간은 각각 2~3주, 2~4주 정도였습니다.
이러한 반응은 대개 용량 감소 또는 이맃틴메실레이트 투여 중단으로 관리할 수 있지만, 영구적으로 투여를 중단해야 할 수도 있습니다.
표 5: 새로 진단된 CML 임상시험에서의 검사실 이상 (이맃틴메실레이트 vs IFN+Ara-C)
|
이맃틴메실레이트 |
IFN+Ara-C |
|||
|
CTC 등급 |
3등급 |
4등급 |
3등급 |
4등급 |
|
혈액학적 지표* |
||||
|
– 호중구감소증* |
13.1 |
3.6 |
20.8 |
4.5 |
| − 혈소판 감소증* | 8.5 | 0.4 | 15.9 | 0.6 |
| − 빈혈 | 3.3 | 1.1 | 4.1 | 0.2 |
| 생화학 매개변수 | ||||
| − 크레아티닌 상승 | 0 | 0 | 0.4 | 0 |
| − 빌리루빈 상승 | 0.9 | 0.2 | 0.2 | 0 |
| − 알칼리성 인산 분해 효소 상승 | 0.2 | 0 | 0.8 | 0 |
| − SGOT (AST)/SGPT (ALT) 상승 | 4.7 | 0.5 | 7.1 | 0.4 |
|
약자: CML, 만성 골수성 백혈병; IFN, 인터페론-알파; SGOT, 혈청 글루탐산 옥살로아세트산 트랜스아미나아제(현재 AST로 지칭); SGPT, 혈청 글루탐산 피루브산 트랜스아미나아제(현재 ALT로 지칭). *p 0.001 미만(두 치료 그룹 간 3등급 또는 4등급 이상반응의 차이) |
||||
표 6: 새로 진단된 CML 임상시험에서 임상적으로 관련 있는 3/4 등급* 실험실 검사 이상반응 발생률(이매티닙 메실레이트 대 닐로티닙)
|
이매티닙 메실레이트 400 mg 1일 1회 복용 N=280 (%) |
닐로티닙 300 mg 1일 2회 복용 N=279 (%) |
|
| 혈액학적 매개변수 | ||
| 혈소판 감소증 | 9 | 10 |
| 호중구 감소증 | 22 | 12 |
| 빈혈 | 6 | 4 |
| 생화학 매개변수 | ||
| 리파아제 상승 | 4 | 9 |
| 고혈당증 | <1 | 7 |
| 저인산혈증 | 10 | 8 |
| 총 빌리루빈 상승 | <1 | 4 |
| SGPT (ALT) 상승 | 3 | 4 |
| 고칼륨혈증 | 1 | 2 |
| 저나트륨혈증 | <1 | 1 |
| 저칼륨혈증 | 2 | <1 |
| SGOT (AST) 상승 | 1 | 1 |
| 알부민 감소 | <1 | 0 |
| 저칼슘혈증 | <1 | <1 |
알칼리 인산 분해 효소 상승
<1
0
크레아티닌 상승
<1
0
약어: CML, 만성 골수성 백혈병; SGOT, 청 글루탐산 옥살로 아세트산 아미노전이효소는 이제 아스파르테이트 아미노전이효소(AST)로 불림; SGPT, 청 글루탐산 피루브산 아미노전이효소는 이제 알라닌 아미노전이효소(ALT)로 불림.
*NCI 일반 독성 용어 기준, 버전 3.0.
표 7: 다른 CML 임상시험에서의 실험실 이상
|
골수모세포증 급성기 |
가속기 |
만성기, IFN 실패 400 mg |
||||
|
CTC 등급(1) |
3등급 |
4등급 |
3등급 |
4등급 |
3등급 |
4등급 |
|
혈액학 파라미터 |
||||||
|
– 중성구 감소증 |
16 |
48 |
23 |
36 |
27 |
9 |
|
– 혈소판 감소증 |
30 |
33 |
31 |
13 |
21 |
<1 |
|
– 빈혈 |
42 |
11 |
34 |
7 |
6 |
1 |
|
생화학 파라미터 |
||||||
|
– 크레아티닌 상승 |
1.5 |
0 |
1.3 |
0 |
0.2 |
0 |
|
– 빌리루빈 상승 |
3.8 |
0 |
2.1 |
0 |
0.6 |
0 |
|
– 알칼리성 인산 분해 효소 상승 |
4.6 |
0 |
5.5 |
0.4 |
0.2 |
0 |
|
– SGOT(AST) 상승 |
1.9 |
0 |
3.0 |
0 |
2.3 |
0 |
|
– SGPT(ALT) 상승 |
2.3 |
0.4 |
4.3 |
0 |
2.1 |
0 |
약어: CML, 만성 골수성 백혈병; CTC, 일반 용어 평가 기준; IFN, 인터페론-알파; SGOT는 이제 아스파르테이트 아미노전이효소(AST)로 불립니다; SGPT는 이제 알라닌 아미노전이효소(ALT)로 불립니다.
(1)CTC 등급: 중성구감소증(등급 3 0.5~1.0×109/L 이상, 등급 4 0.5×109/L 미만), 혈소판감소증(등급 3 10~50×109/L 이상, 등급 4 10×109/L 미만), 빈혈(혈색소 65~80g/L 이상, 등급 4 65g/L 미만), 크레아티닌 상승(등급 3 정상 상한치의 3~6배 이상, 등급 4 정상 상한치의 6배 이상), 빌리루빈 상승(등급 3 정상 상한치의 3~10배 이상, 등급 4 정상 상한치의 10배 이상), 알칼리성 인산분해효소 상승(등급 3 정상 상한치의 5~20배 이상, 등급 4 정상 상한치의 20배 이상), SGOT 또는 SGPT 상승(등급 3 정상 상한치의 5~20배 이상, 등급 4 정상 상한치의 20배 이상).
간 독성
CML 환자의 약 5%에서 중증 아미노전이효소 또는 빌리루빈 상승이 발생했으며(표 6 및 7 참조), 이는 대개 용량 감소 또는 투약 중단(해당 에피소드의 중간 기간은 약 1주일)으로 관리했습니다. 간 검사 수치 이상 때문에 1% 미만의 CML 환자에서 투약을 영구 중단했습니다. 편두통으로 아세트아미노펜을 정기적으로 복용했던 한 환자는 급성 간부전으로 사망했습니다. GIST 2상 시험에서 등급 3 또는 4 SGPT(ALT) 상승이 6.8%의 환자에서, 등급 3 또는 4 SGOT(AST) 상승이 4.8%의 환자에서 관찰되었습니다. 빌리루빈 상승은 2.7%의 환자에서 관찰되었습니다.
소아 집단에서의 이상 반응
단독 요법
연구된 93명의 소아 환자에게 이맘티닙메실레이트를 투여했을 때의 전반적인 안전성 프로파일은 성인 환자에서의 결과와 유사했으나, 근골격계 통증은 더 드물었고(20.5%) 말초부종은 보고되지 않았습니다. 구역과 구토는 성인 환자에서와 비슷한 발생 빈도로 가장 흔하게 보고된 개별 이상 반응이었습니다. 대부분 환자가 언젠가는 이상 반응을 경험했습니다. 모든 이상 반응 유형에 걸쳐 등급 3/4 사례의 발생률은 75%였으며, CML 소아 환자에서 가장 높은 등급 3/4 발생률을 보인 사례는 주로 골수억제와 관련된 것이었습니다.
복합 화학요법과 병용
예상 5년 무사건 생존율(EFS)이 45% 미만으로 매우 높은 위험도를 가진 소아 및 젊은 성인 ALL 환자들이 유도 요법 이후 다기관 비무작위 임상시험에 등록되었습니다. 연구 대상군에는 중간 연령 10세(1~21세)의 환자가 포함되었으며, 그 중 61%는 남성, 75%는 백인, 7%는 흑인, 6%는 아시아/태평양 섬 출신이었습니다. Ph+ ALL 환자(n = 92)는 이맘티닙메실레이트 투여 대상으로 배정되어 5개 연속 코호트에서 치료를 받았습니다. 이맘티닙메실레이트 노출 정도는 연속 코호트마다 체계적으로 증가되었습니다(더 이른 시기 투여 개시 및 더 오랜 기간 투여).
집중 화학요법과 병용한 이맘티닙메실레이트의 안전성은 Ph+ ALL 환자 92명에서 등급 3 및 4 이상 반응(중성구감소증 [750/mcL 미만] 및 혈소판감소증 [75,000/mcL 미만])의 발생률과, 이맘티닙메실레이트를 투여하지 않은 Ph- ALL 환자 65명에서의 발생률을 비교하여 평가했습니다. 또한 이맘티닙메실레이트 투여군과 비투여군의 요법 주기 간 이상 반응 발생률을 비교하여 안전성을 평가했습니다. 이 프로토콜에는 최대 18주기의 요법이 포함되었습니다. 환자들은 총 누적 1,425 주기의 요법을 노출되었으며, 그 중 778 주기는 이맘티닙메실레이트 투여군, 647 주기는 비투여군이었습니다. Ph+ ALL 환자에 비해 Ph- ALL 환자에서 5% 이상 높은 발생률을 보인 이상 반응, 또는 이맘티닙메실레이트 투여 주기에서 1% 이상 더 높은 발생률을 보인 이상 반응은 표 8에 제시되어 있습니다.
|
표 8: 연구 약물 투여 환자 또는 연구 약물 투여 주기에서 더 자주 보고된 이상 반응(5% 이상 또는 1% 이상) |
||||
|
이상 반응 등급 3 및 4 이상 반응 |
환자별 발생률 이맘티닙메실레이트 투여 Ph+ ALL |
환자별 발생률 이맘티닙메실레이트 비투여 Ph- ALL |
환자별/주기별 발생률 이맘티닙메실레이트 투여군* |
환자별/주기별 발생률 이맘티닙메실레이트 비투여군** |
|
N = 92 n (%) |
N = 65 n (%) |
N = 778 n (%) |
N = 647 n (%) |
|
|
구역 및/또는 구토 |
15 (16) |
6 (9) |
28 (4) |
8 (1) |
|
저칼륨혈증 |
31 (34) |
16 (25) |
72 (9) |
32 (5) |
|
폐렴 |
7 (8) |
1 (1) |
7 (1) |
1 (< 1) |
|
흉수 |
6 (7) |
0 |
6 (1) | |
|
복통 |
8 (9) |
2 (3) |
9 (1) |
3 (< 1) |
|
식욕부진 |
10 (11) |
3 (5) |
19 (2) |
4 (1) |
|
출혈 |
11 (12) |
4 (6) |
17 (2) |
8 (1) |
|
저산소증 |
8 (9) |
2 (3) |
12 (2) |
2 (< 1) |
|
근육통 |
5 (5) |
0 |
4 (1) |
1 (< 1) |
|
구내염 |
15 (16) |
8 (12) |
22 (3) |
14 (2) |
|
설사 |
8 (9) |
3 (5) |
12 (2) |
3 (< 1) |
|
발진/피부장애 |
4 (4) |
0 |
5 (1) |
0 |
|
감염 |
49 (53) |
32 (49) |
131 (17) |
92 (14) |
|
간(트랜스아미나제 및/또는 빌리루빈) |
52 (57) |
38 (58) |
172 (22) |
113 (17) |
|
저혈압 |
10 (11) |
5 (8) |
16 (2) |
6 (1) |
|
골수억제 |
||||
|
호중구감소증 (< 750/mcL) |
92 (100) |
63 (97) |
556 (71) |
218 (34) |
|
혈소판감소증 (< 75,000/mcL) |
90 (92) |
63 (97) |
431 (55) |
329 (51) |
|
약어: Ph+ ALL, 필라델피아 염색체 양성 급성 림프구성 백혈병; Ph- ALL, 필라델피아 염색체 음성 급성 림프구성 백혈병. *이마티닙 메실레이트를 포함한 치료 주기당 환자당 이상반응(AE) 발생 빈도로 정의(Ph+ ALL 환자 중 이마티닙 메실레이트가 포함된 주기를 받은 환자 포함). **이마티닙 메실레이트를 포함하지 않은 치료 주기당 환자당 이상반응 발생 빈도로 정의(이마티닙 메실레이트가 포함되지 않은 주기를 받은 Ph+ ALL 환자와 이마티닙 메실레이트를 전혀 투여받지 않은 모든 Ph- ALL 환자 포함). |
||||
다른 하위집단에서의 이상반응
65세 이상의 고령 환자에서 부종을 제외하고는 이상반응 발생률이나 중증도가 증가하지 않았다. 여성에서는 호중구감소증, 1/2도 표재성 부종, 두통, 구역질, 발열, 구토, 발진 및 피로의 빈도가 증가하였다. 인종과 관련된 차이는 발견되지 않았으나 하위집단이 너무 작아 적절한 평가가 어려웠다.
급성 림프구성 백혈병
Ph+ ALL에서의 이상반응은 Ph+ CML과 유사하였다. Ph+ ALL 연구에서 가장 빈번히 보고된 약물 관련 이상반응은 경증 구역질, 구토, 설사, 근육통, 근육 경련 및 발진이었다. 표재성 부종은 모든 연구에서 공통적으로 나타났으며, 주로 안와주위 또는 하지 부종으로 기술되었다. 이러한 부종은 환자의 6.3%에서 3/4도로 보고되었으며, 이뇨제, 기타 보조 조치 또는 일부 환자에서는 이마티닙 메실레이트 용량을 낮추어 치료할 수 있다.
골수형성이상/골수증식성 질환
2상 연구에서 이마티닙 메실레이트로 치료받은 MDS/MPD 환자에게서 최소 10% 이상 보고된 약물 관련 여부와 상관없는 이상반응은 표 9에 나타나 있다.
표 9: 2상 연구에서 MPD 환자에게 보고된 약물 관련 여부와 상관없는 이상반응(최소 한 명 이상의 환자) 전체 등급 (>= 10% 전체 환자)
|
선호 용어 |
N=7 |
|
구역질 |
4 (57.1) |
|
설사 |
3 (42.9) |
|
빈혈 |
2 (28.6) |
|
피로 |
2 (28.6) |
|
근육 경련 |
3 (42.9) |
|
Arthralgia |
2 (28.6) |
|
Periorbital Edema |
2 (28.6) |
|
약어: MPD, 골수증식성 질환. |
|
공격적 전신 비만세포증
모든 공격적 전신 비만세포증(ASM) 환자는 적어도 한 가지 이상의 이상반응을 경험했습니다. 가장 빈번하게 보고된 이상반응은 설사, 구역질, 복수, 근육 경련, 호흡곤란, 피로, 말초부종, 빈혈, 가려움증, 발진 및 하기도 감염이었습니다. 2상 연구에 참여한 ASM 환자 5명 중 이 약물과 관련된 이상반응이나 비정상적인 검사실 수치로 인해 이 약을 중단한 사람은 없었습니다.
과호산구증후군 및 만성 호산구성 백혈병
과호산구증후군/만성 호산구성 백혈병 환자군의 안전성 프로파일은 Ph+ CML과 같은 다른 혈액 악성 종양 환자군에서 관찰된 이 약의 안전성 프로파일과 다르지 않은 것으로 보입니다. 모든 환자들은 적어도 한 가지 이상의 이상반응을 경험했으며, 가장 흔한 것은 위장관, 피부 및 근골격계 장애였습니다. 혈액학적 이상 역시 빈번했으며 CTC 등급 3의 백혈구감소증, 호중구감소증, 림프구감소증 및 빈혈 사례가 있었습니다.
진피 육종종괴
2상 연구에서 진피육종종괴 환자 12명을 이 약으로 치료했을 때 약물과의 관련 여부와 상관없이 10% 이상에서 보고된 이상반응은 표 10과 같습니다.
표 10: 2상 연구에서 진피육종종괴 환자들에게 보고된 모든 등급의 약물 관련 여부와 상관없는 이상반응(10% 이상 전체 환자)
|
선호 용어 |
N=12 |
|
구역질 |
5 (41.7) |
|
설사 |
3 (25.0) |
|
구토 |
3 (25.0) |
|
Periorbital Edema |
4 (33.3) |
|
안면 부종 |
2 (16.7) |
|
발진 |
3 (25.0) |
|
피로 |
5 (41.7) |
|
말초부종 |
4 (33.3) |
|
발열 |
2 (16.7) |
|
안구 부종 |
4 (33.3) |
|
눈물 과다 분비 |
3 (25.0) |
|
운동성 호흡곤란 |
2 (16.7) |
|
빈혈 |
3 (25.0) |
|
비염 |
2 (16.7) |
|
식욕 부진 |
2 (16.7) |
| 약어: DFSP, 진피육종종괴. | |
2상 연구에서 진피육종종괴 환자 12명 중 임상적으로 유의하거나 중증의 실험실 이상 수치는 표 11과 같습니다.
표 11: 2상 연구에서 진피육종종괴 환자들에게 보고된 실험실 이상
|
N=12 |
||
|
CTC 등급(1) |
3등급 % |
4등급 % |
|
혈액학적 수치 |
||
|
– 빈혈 |
17 |
0 |
|
– 혈소판 감소증 |
17 |
0 |
|
– 호중구 감소증 |
0 |
8 |
|
생화학 매개변수 |
||
|
– 크레아티닌 상승 |
0 |
8 |
|
약어: CTC, 일반 용어 기준. (1)CTC 등급: 호중구 감소증(3등급 ≥0.5~1.0×109/L, 4등급 <0.5×109/L), 혈소판 감소증(3등급 ≥10~50×109/L, 4등급 <10×109/L), 빈혈(3등급 ≥65~80g/L, 4등급 <65g/L), 크레아티닌 상승(3등급 >3~6x정상 상한치[ULN], 4등급 >6xULN). |
||
위장관 기질 종양
절제 불가능하고/또는 전이성 악성 GIST
3상 시험에서 이마티닙 메실레이트로 치료받은 대부분의 환자가 언젠가 부작용을 경험했다. 가장 자주 보고된 부작용은 부종, 피로, 구역질, 복통, 설사, 발진, 구토, 근육통, 빈혈, 그리고 식욕부진이었다. 부작용으로 인해 총 89명의 환자(5.4%)에서 투약이 중단되었다. 천안와 부종이나 하지 부종 등의 표재성 부종은 이뇨제, 다른 보조 치료 또는 이마티닙 메실레이트 용량 감소로 관리되었다[용법ㆍ용량 2.13 참조]. 중증(CTC 3/4등급) 부종은 182명의 환자(11.1%)에서 관찰되었다.
시험약과의 관련 여부에 관계없이 이마티닙 메실레이트로 치료받은 환자의 10% 이상에서 보고된 부작용은 표 12에 나와 있다.
전반적으로 모든 등급의 부작용 발생률과 중증 부작용(CTC 3등급 이상)의 발생률은 두 치료군 사이에서 유사했지만 부종은 800mg군에서 더 자주 보고되었다.
표 12: 3상 절제 불가능하고/또는 전이성 악성 GIST 임상시험에서 두 군 중 하나에서 빈도가 10% 이상인 시험약과 무관한 부작용 환자 수(%)(전체 분석군)
|
이마티닙 400mg N = 818 |
이마티닙 800mg N = 822 |
|||||||
|
보고 또는 지정된 용어 |
전체 등급 % |
3/4/5등급 % |
전체 등급 % |
3/4/5등급 % |
||||
|
부종 |
76.7 |
9.0 |
86.1 |
13.1 |
||||
|
피로/권태감, 나른함, 무기력증 |
69.3 |
11.7 |
74.9 |
12.2 |
||||
|
구역질 |
58.1 |
9.0 |
64.5 |
7.8 |
||||
|
복통/경련 |
57.2 |
13.8 |
55.2 |
11.8 |
||||
|
설사 |
56.2 |
8.1 |
58.2 |
8.6 |
||||
|
발진/피부박리 |
38.1 |
7.6 |
49.8 |
8.9 |
||||
|
구토 |
37.4 |
9.2 |
40.6 |
7.5 |
||||
|
근육통 |
32.2 |
5.6 |
30.2 |
3.8 |
||||
|
빈혈 |
32.0 |
4.9 |
34.8 |
6.4 편식증(식욕부진) |
31.1 |
6.6 |
35.8 |
4.7 |
|
기타 위장관 독성 |
25.2 |
8.1 |
28.1 |
6.6 |
||||
|
두통 |
22.0 |
5.7 |
19.7 |
3.6 |
||||
|
기타 통증(종양 관련 통증 제외) |
20.4 |
5.9 |
20.8 |
5.0 |
||||
|
기타 피부/피부 독성 |
17.6 |
5.9 |
20.1 |
5.7 |
||||
|
백혈구감소증 |
17.0 |
0.7 |
19.6 |
1.6 |
||||
|
기타 전신 증상 |
16.7 |
6.4 |
15.2 |
4.4 |
||||
|
기침 |
16.1 |
4.5 |
14.5 |
3.2 |
||||
|
감염(호중구감소증 없음) |
15.5 |
6.6 |
16.5 |
5.6 |
||||
|
가려움증 |
15.4 |
5.4 |
18.9 |
4.3 |
||||
|
기타 신경 독성 |
15.0 |
6.4 |
15.2 |
4.9 |
||||
|
기침 |
16.1 |
4.5 |
14.5 |
3.2 |
||||
|
감염(호중구감소증 없음) |
15.5 |
6.6 |
16.5 |
5.6 |
||||
|
가려움증 |
15.4 |
5.4 |
18.9 |
4.3 |
||||
|
기타 신경 독성 |
15.0 |
6.4 |
15.2 |
4.9 |
||||
|
변비 |
14.8 |
5.1 |
14.4 |
4.1 |
||||
|
기타 신장/비뇨기 독성 |
14.2 |
6.5 |
13.6 |
5.2 |
||||
|
관절통(관절 통증) |
13.6 |
4.8 |
12.3 |
3.0 |
||||
|
호흡곤란(숨가쁨) |
13.6 |
6.8 |
14.2 |
5.6 |
||||
|
호중구감소증(ANC < 1.0 x 109/L) 없이 발열 |
13.2 |
4.9 |
12.9 |
3.4 |
||||
|
발한(식은땀) |
12.7 |
4.6 |
8.5 |
2.8 |
||||
|
기타 출혈 |
12.3 |
6.7 |
13.3 |
6.1 |
||||
|
체중증가 |
12.0 |
1.0 |
10.6 |
0.6 |
||||
|
탈모증 |
11.9 |
4.3 |
14.8 |
3.2 |
||||
|
소화불량/heartburn |
11.5 |
0.6 |
10.9 |
0.5 |
||||
|
호중구감소증/과립구감소증 |
11.5 |
3.1 |
16.1 |
4.1 |
||||
|
오한/발열 |
11.0 |
4.6 |
10.2 |
3.0 |
||||
|
현기증/빛멀미 |
11.0 |
4.8 |
10.0 |
2.8 |
||||
|
크레아티닌 증가 |
10.8 |
0.4 |
10.1 |
0.6 |
||||
|
복부팽만 |
10.0 |
0.2 |
10.1 |
0.1 |
||||
|
구내염/인후염(구강/인두 점막염) |
9.2 |
5.4 |
10.0 |
4.3 |
||||
|
림프구감소증 |
6.0 |
0.7 |
10.1 |
1.9 |
||||
|
약어: ANC, 절대 호중구 수; GI, 위장관; GIST, 위장관 기질종양. |
||||||||
3상 GIST 시험에서 임상적으로 관련성이 있거나 중증의 일상적인 혈액학적 또는 생화학적 검사값의 이상은 보고되거나 평가되지 않았습니다. 2상 GIST 시험에서 보고된 중증의 이상 실험실 값은 표 13에 제시되어 있습니다.
표 13: 2상 절제 불가능 및/또는 전이성 악성 GIST 시험에서의 실험실 이상
|
400 mg (n = 73) % |
600 mg (n = 74) % |
|||
|
CTC 등급1 |
3등급 |
4등급 |
3등급 |
4등급 |
|
혈액학적 지표 |
||||
|
− 빈혈 |
3 |
0 |
8 |
1 |
|
− 혈소판감소증 |
0 |
0 |
1 |
0 |
|
− 호중구감소증 |
7 |
3 |
8 |
3 |
|
생화학적 지표 |
||||
|
− 크레아티닌 상승 |
0 |
0 |
3 | |
|
6 부작용 |
0 |
|||
|
– 알부민 감소 |
3 |
0 |
4 |
0 |
|
– 빌리루빈 증가 |
1 |
0 |
1 |
3 |
|
– 알칼리성 인산분해효소 증가 |
0 |
0 |
3 |
0 |
|
– SGOT (AST) 증가 |
4 |
0 |
3 |
3 |
|
– SGPT (ALT) 증가 |
6 |
0 |
7 |
1 |
|
약어: CTC, 일반 용어 기준; GIST, 위장관 기질 종양; SGOT, 혈청 글루타믹-옥살로아세트산 트랜스아미나아제는 현재 아스파르테이트 아미노전이효소(AST)로 불림; SGPT, 혈청 글루타믹-피루빈산 트랜스아미나아제는 현재 알라닌 아미노전이효소(ALT)로 불림. 1CTC 등급: 호중구감소증(3등급 0.5 × 109/L 이상, 1.0 × 109/L 미만, 4등급 0.5 × 109/L 미만), 혈소판감소증(3등급 10 × 109/L 이상, 50 × 109/L 미만, 4등급 10 × 109/L 미만), 빈혈(3등급 65g/L 이상, 80g/L 미만, 4등급 65g/L 미만), 크레아티닌 증가(3등급 정상 상한치의 3배 초과 6배 이하, 4등급 정상 상한치의 6배 초과), 빌리루빈 증가(3등급 정상 상한치의 3배 초과 10배 이하, 4등급 정상 상한치의 10배 초과), 알칼리성 인산분해효소, SGOT 또는 SGPT 증가(3등급 정상 상한치의 5배 초과 20배 이하, 4등급 정상 상한치의 20배 초과), 알부민(3등급 20g/L 미만). |
||||
GIST 보조 치료
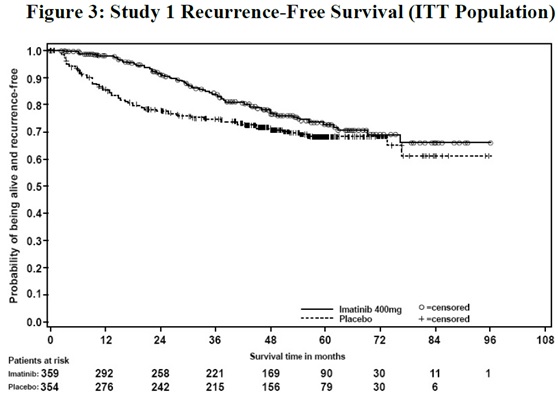
연구 1에서 이마티닙 메실레이트 및 위약 치료 환자 대부분은 적어도 한 번 이상 부작용을 경험했습니다. 가장 빈번히 보고된 부작용은 다른 임상 연구에서 다른 환자군에서 보고된 것과 유사한데, 설사, 피로, 구역, 부종, 헤모글로빈 감소, 발진, 구토 및 복통이 포함됩니다. 이전에 다른 환자군(절제 불가능 및/또는 전이성 악성 GIST 환자 포함)에서 보고되지 않은 새로운 부작용은 GIST 보조 치료 환경에서 보고되지 않았습니다. 부작용으로 인해 이마티닙 메실레이트 투여군 57명(17%)과 위약 투여군 11명(3%)에서 약물 투여가 중단되었습니다. 중단 시점에서 가장 빈번히 보고된 부작용은 부종, 소화기계 장애(구역, 구토, 복부 팽만 및 설사), 피로, 헤모글로빈 저하, 발진이었습니다.
연구 2에서 부작용으로 인한 투약 중단은 이마티닙 메실레이트 12개월 치료군 15명(8%)과 36개월 치료군 27명(14%)에서 발생했습니다. 이전 시험과 마찬가지로 가장 흔한 부작용은 설사, 피로, 구역, 부종, 헤모글로빈 감소, 발진, 구토, 복통이었습니다.
연구 약물과 관계없이 이마티닙 메실레이트로 치료받은 환자의 5% 이상에서 보고된 부작용은 표 14(연구 1)와 표 15(연구 2)에 나와 있습니다. 두 시험 모두에서 이마티닙 메실레이트 치료와 관련된 사망 사례는 없었습니다.
표 14. 연구 1에서 보고된 이마티닙 메실레이트 치료군 5% 이상에서 발생한 연구 약물과 관계없는 부작용(1)
|
바람직하지 않은 용어 |
모든 CTC 등급 |
CTC 3* 등급 이상 |
||
|
이마티닙 메실레이트 (n = 337) |
위약 (n = 345) |
이마티닙 메실레이트 (n = 337) |
위약 (n = 345) |
|
|
% |
% |
% |
% |
|
|
설사 |
59.3 |
29.3 |
3.0 |
1.4 |
|
피로 |
57.0 |
40.9 |
2.1 |
1.2 |
|
구역 |
53.1 |
27.8 |
2.4 |
1.2 |
|
안와주위 부종 |
47.2 |
14.5 |
1.2 |
0 |
|
헤모글로빈 감소 |
46.9 |
27.0 |
0.6 |
0 |
|
말초부종 |
26.7 |
14.8 |
0.3 |
0 |
|
발진 (박리성) |
26.1 |
12.8 |
2.7 |
0 |
|
구토 |
25.5 |
13.9 |
2.4 |
0.6 |
|
복통 |
21.1 |
22.3 |
3.0 |
1.4 |
|
두통 |
19.3 |
20.3 |
0.6 |
0 |
|
소화불량 |
17.2 |
13.0 |
0.9 |
0 |
|
식욕부진 |
16.9 |
8.7 |
0.3 |
0 |
|
체중증가 |
16.9 |
11.6 |
0.3 |
0 |
|
ALT 증가 |
16.6 |
13.0 |
2.7 |
0 |
|
근육경련 |
16.3 |
3.3 |
0 |
0 |
|
호중구 감소 |
16.0 |
6.1 |
3.3 |
0.9 |
|
관절통 |
15.1 |
14.5 |
0 |
0.3 |
|
백혈구 감소 |
14.5 |
4.3 |
0.6 |
0.3 |
|
변비 |
12.8 |
17.7 |
0 |
0.3 |
|
현기증 |
12.5 |
10.7 |
0 |
0.3 |
|
AST 증가 |
12.2 |
7.5 |
2.1 |
0 |
|
근육통 |
12.2 |
11.6 |
0 |
0.3 |
|
혈중 크레아티닌 증가 |
11.6 |
5.8 |
0 |
0.3 |
|
기침 |
11.0 |
11.3 |
0 |
0 |
|
가려움증 |
11.0 |
7.8 |
0.9 |
0 |
|
체중 감소 |
10.1 |
5.2 |
0 |
0 |
|
고혈당증 |
9.8 |
11.3 |
0.6 |
1.7 |
|
불면증 |
9.8 |
7.2 |
0.9 |
0 |
|
눈물 분비 증가 |
9.8 |
3.8 |
0 |
0 |
|
탈모 |
9.5 |
6.7 |
0 |
0 |
|
복통 |
8.9 |
9.6 |
0 |
0 |
|
발진 |
8.9 |
5.2 |
0.9 |
0 |
|
복부 팽만감 |
7.4 |
6.4 |
0.3 |
0.3 |
|
등 통증 |
7.4 |
8.1 |
0.6 |
0 |
|
근육 통증 |
7.4 |
7.2 |
0.3 |
0 |
|
저칼륨혈증 |
7.1 |
2.0 |
0.9 |
0.6 |
|
우울증 |
6.8 |
6.4 |
0.9 |
0.6 |
|
얼굴 부종 |
6.8 |
1.2 |
0.3 |
0 |
|
혈알칼리성 인산효소 증가 |
6.5 |
7.5 |
0 |
0 |
|
건조한 피부 |
6.5 |
5.2 |
0 |
0 |
|
미각 이상 |
6.5 |
2.9 |
0 |
0 |
|
상복부 통증 |
6.2 |
6.4 |
0.3 |
0 |
|
말초 신경병증 |
5.9 |
6.4 |
0 |
0 |
|
저칼슘혈증 |
5.6 |
1.7 |
0.3 |
0 |
|
백혈구 감소 |
5.0 |
2.6 |
0.3 |
0 |
|
혈소판 감소 |
5.0 |
3.5 |
0 |
0 |
|
구내염 |
5.0 |
1.7 |
0.6 |
0 |
|
상기도 감염 |
5.0 |
3.5 |
0 |
0 |
|
시력 흐림 |
5.0 |
2.3 |
0 |
0 |
|
약어: CTC, 일반 용어 기준; GIST, 위장관 기질종양; SGOT, 혈청 글루탐산 옥살아세트산 트랜스아미나제는 이제 아스파르테이트 아미노전이효소(AST)로 지칭됩니다; SGPT, 혈청 글루탐산-피루빈산 트랜스아미나제는 이제 알라닌 아미노전이효소(ALT)로 불립니다. *NCI 일반 용어 기준 부작용, 버전 3.0. (1)치료와 관련 여부와 상관없이 환자의 5% 이상에서 발생한 모든 부작용이 나열되었습니다. 동일한 부작용이 반복하여 발생한 환자는 해당 부작용 범주에서 한 번만 계산됩니다. |
||||
표 15: 치료 약물 관계 없이 선호 용어별 등급 및 3/4등급 부작용(이매티닙 메실레이트 치료 환자 5% 이상) 연구 2(1)
|
선호 용어 |
전체 CTC 등급 |
CTC 3등급 및 상위 |
||
|
이매티닙 메실레이트 12개월 (N = 194) |
이매티닙 메실레이트 36개월 (N = 198) |
이매티닙 메실레이트 12개월 (N = 194) |
이매티닙 메실레이트 36개월 (N = 198) |
|
|
% |
% |
% |
% |
|
|
1건 이상의 이상반응이 있는 환자 |
99.0 |
100.0 |
20.1 |
32.8 |
|
헤모글로빈 감소 |
72.2 |
80.3 |
0.5 |
0.5 |
|
안구주위부종 |
59.3 |
74.2 |
0.5 |
1.0 |
|
혈중 젖산 탈수소효소 증가 |
43.3 |
60.1 |
0 |
0 |
|
설사 |
43.8 |
54.0 |
0.5 |
2.0 |
|
구역질 |
44.8 |
51.0 |
1.5 |
0.5 |
|
근육 경련 |
30.9 |
49.0 |
0.5 |
1.0 |
|
피로 |
48.5 |
48.5 |
1.0 |
0.5 |
|
백혈구 수 감소 |
34.5 |
47.0 |
2.1 |
3.0 |
|
통증 |
25.8 |
45.5 |
1.0 |
3.0 |
|
혈중 크레아티닌 증가 |
30.4 |
44.4 |
6. 부작용 |
|
|
말초부종 |
33.0 |
40.9 |
0.5 |
1.0 |
|
피부염 |
29.4 |
38.9 |
2.1 |
1.5 |
|
아스파르테이트 아미노전이효소 증가 |
30.9 |
37.9 |
1.5 |
3.0 |
|
알라닌 아미노전이효소 증가 |
28.9 |
34.3 |
2.1 |
3.0 |
|
호중구 감소 |
24.2 |
33.3 |
4.6 |
5.1 |
|
저단백혈증 |
23.7 |
31.8 |
0 |
0 |
|
감염 |
13.9 |
27.8 |
1.5 |
2.5 |
|
체중 증가 |
13.4 |
26.8 |
0 |
0.5 |
|
가려움증 |
12.9 |
25.8 |
0 |
0 |
|
복부팽만 |
19.1 |
24.7 |
1.0 |
0.5 |
|
구토 |
10.8 |
22.2 |
0.5 |
1.0 |
|
소화불량 |
17.5 |
21.7 |
0.5 |
1.0 |
|
저알부민혈증 |
11.9 |
21.2 |
0 |
0 |
|
부종 |
10.8 |
19.7 |
0 |
0.5 |
|
복부팽만 |
11.9 |
19.2 |
0.5 |
0 |
|
두통 |
8.2 |
18.2 |
0 |
0 |
|
눈물증가 |
18.0 |
17.7 |
0 |
0 |
|
관절통 |
8.8 |
17.2 |
0 |
1.0 |
|
알칼라인 인산분해효소 증가 |
10.8 |
16.7 |
0 |
0.5 |
|
호흡곤란 |
6.2 |
16.2 |
0.5 |
1.5 |
|
근육통 |
9.3 |
15.2 |
0 |
1.0 |
|
혈소판 수치 감소 |
11.3 |
14.1 |
0 |
0 |
|
혈중 빌리루빈 증가 |
11.3 |
13.1 |
0 |
0 |
|
Dysgeusia |
9.3 |
12.6 |
0 |
0 |
|
Paresthesia |
5.2 |
12.1 |
0 |
0.5 |
|
시력 흐림 |
10.8 |
11.1 |
1.0 |
0.5 |
|
탈모 |
11.3 |
10.6 |
0 |
0 |
|
식욕 감퇴 |
9.8 |
10.1 |
0 |
0 |
|
변비 |
8.8 |
9.6 |
0 |
0 |
|
발열 |
6.2 |
9.6 |
0 |
0 |
|
우울증 |
3.1 |
8.1 |
0 |
0 |
|
복통 |
2.6 |
7.6 |
0 |
0 |
|
결막염 |
5.2 |
7.6 |
0 |
0 |
|
광과민 반응 |
3.6 |
7.1 |
0 |
0 |
|
어지러움 |
4.6 |
6.6 |
0.5 |
0 |
|
출혈 |
3.1 |
6.6 |
0 |
0 |
|
건조한 피부 |
6.7 |
6.1 |
0.5 |
0 |
|
Nasopharyngitis |
1.0 |
6.1 |
0 |
0.5 |
|
Palpitations |
5.2 |
5.1 |
0 |
0 |
|
약어: AE, 부작용; CTC, 공통 용어 기준. 부작용이 여러 번 발생한 환자는 해당 부작용 범주에서 한 번만 계산됩니다. |
||||
여러 임상시험에서의 부작용
심장 장애:
추정 1%~10%: 심계항진, 심낭 삼출
추정 0.1%~1%: 울혈성 심부전, 빈맥, 폐부종
추정 0.01%~0.1%: 부정맥, 심방 세동, 심장 정지, 심근경색, 협심증
혈관 장애:
추정 1%~10%: 홍조, 출혈
추정 0.1%에서 1%: 고혈압, 저혈압, 말초 부분의 냉기, 레이노 현상, 혈종, 경막하 혈종
조사:
추정 1%에서 10%: 혈중 크레아틴 인산키나제 (CPK) 증가, 혈중 아밀라제 증가
추정 0.1%에서 1%: 혈중 락테이트 탈수소효소 (LDH) 증가
피부 및 피하 조직 장애:
추정 1%에서 10%: 건조한 피부, 탈모, 얼굴 부종, 홍반, 빛에 민감한 반응, 손톱 이상, 자색반점
추정 0.1%에서 1%: 각질박리성 피부염, 물집성 발진, 건선, 농포성 발진, 멍, 땀 증가, 두드러기, 멍에 민감한 경향 증가, 탈모, 피부 색소 결핍, 피부 색소 과다, 손톱 파열, 모낭염, 점막출혈, 다발성 홍반, 지방층염(홍색 결절 포함)
추정 0.01%에서 0.1%: 수포성 발진, 스티븐스-존슨 증후군, 급성 일반화성 농포성 발진, 급성 열성 중성구성 피부병증 (수위트 증후군), 손톱 변색, 혈관신경성 부종, 백혈구파괴성 혈관염
위장관 장애:
추정 1%에서 10%: 복부 팽만, 위식도 역류, 입마름, 위염
추정 0.1%에서 1%: 위궤양, 구내염, 구내궤양, 트림, 검은 변, 식도염, 복수, 헤메토메시스, 입술 염증, 삼키기 곤란, 췌장염
추정 0.01%에서 0.1%: 대장염, 장폐색, 염증성 장질환
일반 장애 및 투여 부위 조건:
추정 1%에서 10%: 약해짐, 부종, 오한
추정 0.1%에서 1%: 피로감
혈액 및 림프계 장애:
추정 1%에서 10%: 전혈소판감소증, 열성 중성구감소증, 림프구감소증, 과민증
추정 0.1%에서 1%: 혈소판증가증, 골수억제, 림프절염
추정 0.01%에서 0.1%: 용혈성 빈혈, 혈소판감소증
간담도 장애:
추정 0.1%에서 1%: 간염, 황달
추정 0.01%에서 0.1%: 간기능부전 및 간괴사1
면역계 장애:
추정 0.01%에서 0.1%: 혈관부종
감염 및 기생충 감염:
추정 0.1%에서 1%: 패혈증, 단순 헤르페스, 대상포진, 세균성 경피염, 요로감염, 위장염
추정 0.01%에서 0.1%: 곰팡이 감염
대사 및 영양 장애:
추정 1%에서 10%: 체중 감소, 식욕 감소
추정 0.1%에서 1%: 탈수, 통풍, 식욕 증가, 고요산혈증, 고칼슘혈증, 고혈당증, 저나트륨혈증, 고칼륨혈증, 저마그네슘혈증
근골격 및 결합 조직 장애:
추정 1%에서 10%: 관절 부종
추정 0.1%에서 1%: 관절 및 근육 경직, 근력 약화, 관절염
신경계/정신 장애:
추정 1%에서 10%: 저림, 감각 저하
추정 0.1%에서 1%: 실신, 말초 신경병증, 졸음, 편두통, 기억력 저하, 성욕 감소, 싸이어티카, 긴장 다리 증후군, 떨림
추정 0.01%에서 0.1%: 뇌압 증가1, 혼동 상태, 경련, 시신경염
신장 및 비뇨기 장애:
추정 0.1%에서 1%: 급성 신부전, 배뇨 빈도 증가, 혈뇨, 신장 통증
생식기 및 유방 장애:
추정 0.1%에서 1%: 유방 확대, 과다월경, 성기능 장애, 남성 유방증, 발기 부전, 월경 불규칙, 유두 통증, 고환 부종
호흡기, 흉부 및 매개재 장애:
추정 1%에서 10%: 코피
추정 0.1%에서 1%: 흉막 삼출
추정 0.01%에서 0.1%: 조직간 폐렴, 폐섬유증, 흉막 통증, 폐고혈압, 폐출혈
내분비 장애:
추정 0.1%–1%: 갑상선 기능 저하, 갑상선 기능 증가
눈, 귀 및 전정 장애:
추정 1%에서 10%: 결막염, 시력 흐림, 안와 부종, 결막 출혈, 건조한 눈
추정 0.1%에서 1%: 어지러움, 이명, 눈 자극, 눈 통증, 공막 출혈, 망막 출혈, 눈꺼풀 염증, 맥락막 부종, 청력 손실, 백내장
추정 0.01%에서 0.1%: 시신경부종1, 녹내장
1일부 사망 사례 포함.